
Boundary conditions in atomistic simulations for extended defects in solids
Christopher Weinberger 1, Lucas Hale 2 and Ian Bakst 1
1 Department of Mechanical Engineering and Mechanics, Drexel University
2 Materials Science and Engineering Division, NIST
Introduction
When we agreed to lead this discussion, our thoughts on this journal club topic was to discuss boundary conditions in atomistic simulations for dislocations but we ended up writing extended defects with the hope that the discussion would lead us in a broader direction than the one we will set directly on "paper". We have been surprised at how much this problem has plagued us in our careers as we have had to revisit the idea of boundary conditions many times. With that said, we feel that it is also important to address what otherwise might seem an old or narrow topic.
Increasing the dimensions of an atomic system lessen the influence that the boundaries have on the structure and behavior of the defects. With the increase in computational power, one might assume that the issues associated with boundary conditions is going away. Instead, it is becoming more and more relevant as researchers are constantly turning to more accurate (and expensive) atomic models, including density functional theory. As such, this problem will continue to be relevant for some time.
Let’s approach this topic in a general sense in that we want to simulate the properties of an infinitely straight dislocation in a crystalline solid using an atomic description. In this vein, we are usually interested in probing either the properties of the dislocation core (its structure and/or its core energy) or the response of the dislocation to externally applied stress fields. Any classical boundary condition will lead to interactions with the dislocation that may influence the observed results. Here, we will discuss the two broad categories of boundary conditions that have seen the most usefulness in these simulation studies: periodic boundary conditions, and continuum-based boundary conditions.
Periodic Boundary Conditions (Dipole Systems)
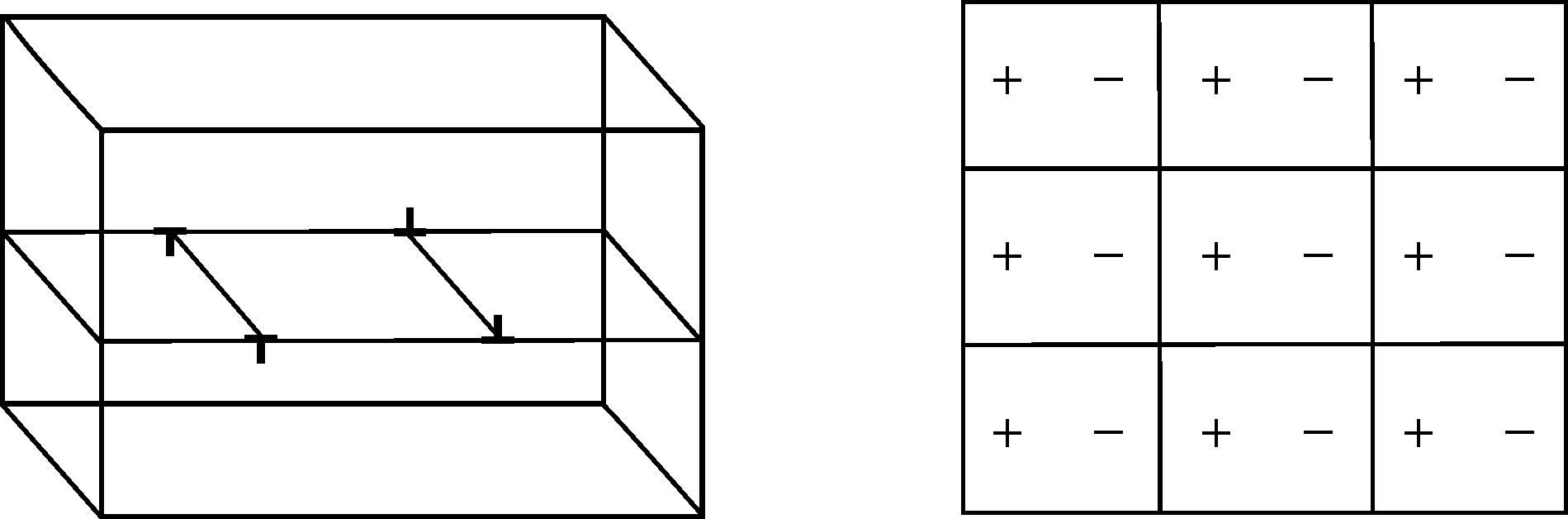
Periodic boundary conditions (PBCs) are probably the most common boundary condition used in atomistic simulations. However, the use of PBCs in simulating infinitely straight dislocations can be quite challenging. If we insist on using periodic boundary conditions in 3 dimensions (which is often done especially in DFT simulations) then we are geometrically limited to systems where the cumulative Burgers vector is zero, i.e. some dipole configuration, as shown in Figure 1. Additionally, care must be taken to account for the interaction between the dislocations, both in constructing a stable system and analyzing results.
Figure 1: Illustration of a simple dislocation dipole in a rectangular simualtion box.
To extract continuum descriptions, it is necessary to compare the atomistically computed energies using a continuum model of a dipole under periodic boundary conditions and this is where the issues arise. The energy in the continuum solution, which arises as a two dimensional infinite sum over the images, is conditionally convergent. This is well described in the papers by Cai et al [1,2] and several books [3,4].
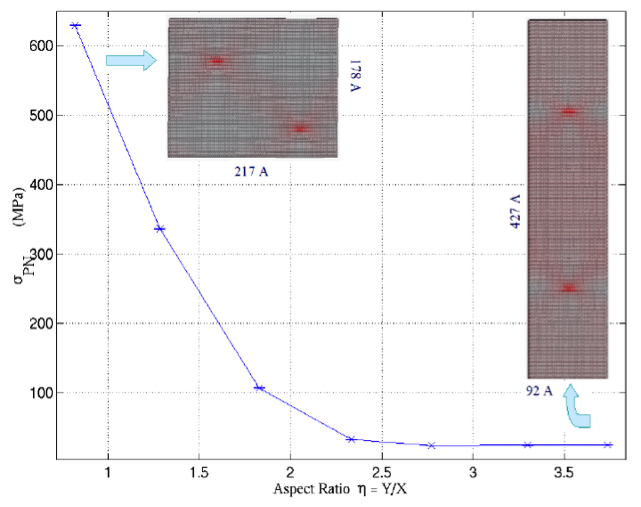
Similarly, the Peierls stress (minimum stress required to move an isolated dislocation) can be estimated by applying a shear to the periodic box. In this setup, the dislocations are preferably oriented such that they do not share a glide plane. The box can be consecutively sheared with subsequent atomic relaxations until the dislocation moves. This critical stress can then be related to the Peierls stress with when the box is sufficiently large. However, caution must be used as the Peierls stress can be dependent on the size of the simulation box, giving erroneous results especially if the Peierls stress is low and the dislocation box size is small [5].
Figure 2: The convergence of the Peierls stress in BCC moly as a function of the box size, from [5].
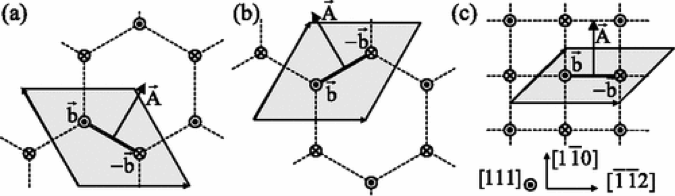
While increasing the simulation cell is possible in classical atomistic simulations using PBCs, such an approach will fail when using electronic structure density functional theory (DFT) in that it is unlikely that the cell size can be increased to a sufficient size. Thus, it has been of particular interest to devise simulation cells that minimize PBC interaction effects as well as the core effects. This has been extensively investigated in [6,7,8]. One of the interesting findings of this work is that the quadrapole arrangement shown in Figure 3(c) does a good job of minimizing the core effects and appears to be the most suitable arrangement for extracting dislocation properties from periodic DFT simulations.
Figure 3: Three different dipole geometries considere for studying dislocation properteis of BCC metals using DFT, from [6].
Continuum-Based Boundary Conditions (Monopole Systems)
While extensive work has been done in extracting information from simulations of dislocations within periodic boundary conditions, it would appear that simulating a single dislocation in an infinite medium would be a more direct method of extracting this information. However, enforcing these boundary conditions, especially in the case of a potentially moving dislocation, can be non trivial.
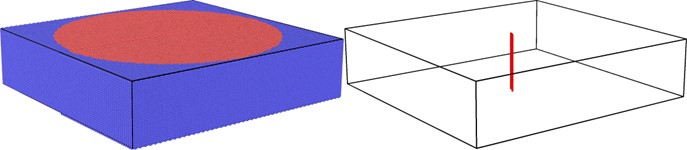
The simplest, and most common method to treat a dislocation monopole is to create the dislocation by displacing the atomic positions for the anisotropic elasticity solution, which is solved using the Stroh formalism [9]. In this method, the atoms inside a radius R are allowed to relax according to the atomic forces while those outside this region are held fixed at their unrelaxed elastic solution positions. This is an almost ideal arrangement as it gives a continuum-based solution for the boundary. However, incompatibility forces will develop at the interface between the relaxed and fixed regions.
Figure 4: An example of a dislocation monopole system. (a) Red atoms are in the active atomistic region, while blue atoms are in the boundary region. (b) Atoms only at the dislocation are shown.
These fixed-boundary monopole systems have been used extensively in the literature to simulate the motion of isolated dislocations by applying uniform strain states to the systems. Of particular note is the extensive use in BCC metals to characterize the non-Schmid effects and we point the reader to two papers (of many) where this has been done [10,11]. This method works well for interatomic potentials where the domain can be modest in size for dislocations. In doing this analysis, care has to be taken as larger incompatibilities will form as the dislocation moves closer to the boundary, and large applied stresses/strains may exceed the limits of linear elasticity.
The application of this boundary condition, the fixed infinite boundary condition, has not been used extensively in DFT studies of dislocations with one exception [6]. The size of the simulation cell that is tractable with DFT simulations is too small to accurately simulate an isolated dislocation as the boundary region is too close to the dislocation resulting in large incompatibility forces. Instead, different boundary conditions have been implemented that retain consistency with continuum-based long range fields, while also allowing for the atoms in the boundary to relax.
The most commonly used technique is based on the classic flexible Greens function boundary condition. The setup is similar to the fixed boundary condition except that an additional transition region is defined at the interface of the atomic and boundary regions. The lattice Greens function is used to calculate the displacements of all atoms associated with negating the atomic forces acting on the atoms in the transition region. An iterative solution is used between relaxing the atomistic region and the transition region until equilibrium is reached across the regions.
This type of BC has been used to study isolated dislocations in BCC metals [10,11] and to test their response under stress. The use of this method, however, requires the implementation of the complicated boundary condition as well as the determination of the lattice Greens function for the lattice of interest.
Related to these types of dislocation studies, there have been successful attempts at pairing of DFT and classical atomistics [12]. This allows for the high accuracy near the core with the ab initio model, and the longer-range elastic behaviors (and larger system sizes) with the classical potential. Note that with this method, it is important that the lattice and elastic constants associated with these different regions are consistent.
Discussion and Outlook
Simulating dislocations in atomistic simulations present a challenging problem for material scientists, mechanicians, and mathematicians. In the case of classical atomistics, if the potential is easy enough to create very large systems, a number of boundary conditions will work (even some not discussed here). For the more computationally time consuming DFT, there are a number of theoretical methods using both PBC and Green’s function boundary condition methods that may result in answers with minimal error. As pointed out in our introduction, the improvement with computer speed will help but will also challenge researchers to use more reliable and expensive methods and thus the problem of boundary condition for dislocations has be come more important perhaps, not less.
It is also worth pointing out that we have ignored the possibility of fixed boundary conditions and free surface boundary conditions. These boundary conditions are usually worse for simulating the properties of dislocations than fixing the boundary using the infinite body solution and are generally avoided unless one is specifically interested in interactions with free or rigid surfaces.
For those who are interested in using these methods, the standardization of these boundary conditions does remain a problem. There are a number of useful and openly available tools known to the authors that are worth checking out. Adding any other tools to the discussion would be much appreciated as well.
Dallas Trinkle has released a code that implements the flexible Greens function boundary condition in VASP [http://dtrinkle.matse.illinois.edu/dokuwiki/doku.php?id=downloads].
For evaluating the Madelung sum in evaluating the properties of dislocation dipoles under periodic boundary conditions, Wei Cai has made a code freely available to do this as well [ http://micro.stanford.edu/~caiwei/Forum/ ].
However, there appears to be limited tools to actually create dislocations in atomistic simulations. As of very recently (March 31st), NIST has released a Code for generating dislocations using the Stroh method is freely available in the atomman Python package being developed as part of NIST’s Interatomic Potential Repository project [https://github.com/usnistgov/atomman/blob/master/Notebooks/atomman.defect.Stroh.ipynb]. The code currently supports saving atomic configurations in LAMMPS data and dump formats. We are sure at this point that we have missed contributions from other authors and we apologize for these oversights.
We would like to point out that there is a lot of potential work out there to be done on these types of boundary conditions. One that comes to mind is in the area of error analysis and uncertainty quantification, especially for those who are more mathematically minded. Several of the papers referenced below mention error associated with dipole configurations and differences between the dipole and infinite boundary conditions. However, we think it would be beneficial to have more rigorous discussions on the types and magnitudes of the errors involved, and what they depend on.
To take this discussion back, we again note that we have only considered dislocations, and infinite straight dislocations at that. One area that could spark some good discussion is in what other types of extended defects should we be considering unique boundary conditions for and what approaches do we need?
References:
[1] Wei Cai, V. V. Bulatov, J. Chang, J. Li, and S. Yip, "Anisotropic Elastic Interactions of a Periodic Dislocation Array", Physical Review Letters, 86, 5727 (2001)
[2] Wei Cai, V. V. Bulatov, J. Chang, J. Li, and S. Yip, "Periodic Image Effects in Dislocation Modeling", Philosophical Magazine A, 83, 539 (2003).
[3] Vasily V. Bulatov and Wei Cai, Computer Simulations of Dislocations, Oxford University Press, Oct 2006.
[4] Wei Cai, "Modelling Dislocations using a Periodic Supercell", Handbook of Materials Modelling, S. Yip, ed. Section 2.21, Springer (2005).
[5] J. Chang, PhD Thesis MIT (2004) : https://dspace.mit.edu/bitstream/handle/1721.1/16601/55011166-MIT.pdf?s…
[6] Ventelon, Lisa, and F. Willaime. "Core structure and Peierls potential of screw dislocations in α-Fe from first principles: cluster versus dipole approaches." Journal of Computer-Aided Materials Design 14.1, 85-94 ((2007).
[7] Clouet, Emmanuel, Lisa Ventelon, and F. Willaime. "Dislocation core field. II. Screw dislocation in iron." Physical Review B 84.22, 224107 (2011).
[8] Clouet, Emmanuel, Lisa Ventelon, and François Willaime. "Dislocation core energies and core fields from first principles." Physical review letters 102: 055502 (2009).
[9] A.N. Stroh, Philosophical Magazine, 3, 625 (1958), A.N. Stroh, J Math Phys Camb, 41, 77 (1962).
[10] Gröger, R., A. G. Bailey, and V. Vitek. "Multiscale modeling of plastic deformation of molybdenum and tungsten: I. Atomistic studies of the core structure and glide of 1/2< 111> screw dislocations at 0K." Acta Materialia 56, 5401 (2008).
[11] Duesbery, M. and-S., and V. Vitek. "Plastic anisotropy in bcc transition metals." Acta Materialia 46, 1481 (1998) .
[12] Tavazza, F., Wagner, R., Chaka, A.M., Levine, L.E. “Vacancy formation energy near an edge dislocation: A hybrid quantum-classical study” Materials Science and Engineering A 400–401 (2005) 72–75.
| Attachment | Size |
|---|---|
| Dislocation Dipole in PBCs | 4.15 KB |
| dipole_pbc_shear.png | 192.97 KB |
| dft_dipole.png | 66.8 KB |
| monopole.jpeg | 20.88 KB |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Dear Christopher,
Dear Christopher,
Thanks for bringing this one up. It is remarkable that this aspect is not as much discussed
in literature as you have pointed out. Especially for those interested in multiscale modeling, it is important to remember the underlying boundary conditions and its effects on dislocation dynamics for example.
I was wondering though if you could also comment if for determining properties of individual dislocations like an Peierls stress of edge dislocation, if it is sufficient to use periodic boundary conditions along the dislocation line direction only?
Because once you introduce an edge dislocation or say a mixed dislocation it would result in a step on the surface of the crystal. If one has a sufficiently large box, would the Peierls stress be reliable?
-Thanks