
The molecular building blocks of a cell include:
♦ membrane components (e.g. fatty acids and phospholipids)
♦ biopolymers (e.g. proteins)
♦ genetic blueprint (e.g. DNA and RNA)
In a previous issue of the journal club, John Dolbow has discussed computational mechanics of biomembrane. I would like to discuss the mechanics issues of proteins and DNA (RNA) from an analytical perspective.
Proteins are linear chains of amino acids and they are the principal component of cytoskeleton and also present in most membranes. Proteins are important functional elements of the cell, where in many cases their structural or mechanical functions are just as important as their biochemical roles. Proteins in the cytoskeleton can form a system of scaffolding to maintain cell integrity; in addition, many fundamentally important biological processes rely on the mechanical response of proteins and their assemblies, including mechanotransduction and molecular motors. The first figure on the left is the molecular structure of mechanosensitive channel of small conductance in Escherichia Coli, which is taken from Orientations of Proteins in Membranes (OPM) database.
{kind=link}
All-atom simulations of the conformational changes of proteins can be prohibitively expensive, which often require the use of supercomputers. Meanwhile, many previous analytical studies were based on thermodynamics. In mechanotransduction for example, Markin and Sachs, and Wiggins and Philips, have developed thermodynamics models to relate the probability of gating of the mechanosensitive channels to membrane properties. Although these theoretical models provide interesting insight into the common features of mechanosensitivity, their validity for any specific system is difficult to judge because the models lack structural details. Moreover, key parameters in these models are not usually obtained from detailed simulations or experiments.
Since structural details and their evolutions are often key to mechanochemical processes, the normal mode analysis (NMA) appears to be quite attractive for studying the thermomechanical properties of various molecular structures at the atomic level. The superpositions of low-frequency normal modes can well describe the global pattern of the structural change (i.e. collective motions) of a biomolecule upon thermal or small mechanical perturbations. The NMA is based on the diagonalization of the 3N x 3N Hessian matrix, i.e. the second derivatives of the mass weighted energy matrix (similar to the global stiffness matrix in finite element method), where N is the number of atoms in the system. Obtaining the Hessian matrix, however, can be quite time consuming due to the involvement of complex empirical atomic potentials during the energy minimization process. A very promising analytical solution to circumvent such difficulty is the elastic-network model (ENM), first proposed by Monique M. Tirion, and this paper is the first one under discussion in the present issue of journal club:
In this paper, a single-parameter harmonic spring is used to model all pairwise interactions between neighboring atoms (as long as they are within a specified cut-off distance). As far as the low-frequency normal modes are of concern, the ENM results are in agreement with those computed from the full atomic model. If only the eigen vectors are of interest, the spring constant can be arbitrary, and the only relevant parameter would be the cut-off distance (typically 0.8-1nm). The ENM is inherently simple, analytical, and of very low computational cost. Moreover, the model may be further simplified by incorporating only representative atoms in the elastic spring network (e.g. Cα), which is widely adopted. The refinement can also be easily done by assigning different spring constants to different components of a protein.
To effectively describe large scale deformations of proteins, a simple approach based on the ENM is of interest, which is the second paper under discussion:
In this paper, based on the Hessian matrix obtained from ENM, displacement "boundary conditions" are applied to certain atoms and through a simple elastic analysis, the corresponding displacements of all atoms in the ENM can be obtained in closed form (Eq. (6) in this paper). To adapt large deformation, such analysis may be carried out repetitively through small increment of perturbations. The paper focused on three families of motor proteins and the approach can be readily extended to other biomolecules. Furthermore, the first author of this paper maintains a website, which is a free web server for computing the eigenmodes of an atomic structure.
An example of a simple ENM exercise, the conformational changes of mechanosensitive channel of large conductance in mycobacterium tuberculosis, is given in the second figure on the left side. Here, the protein subjects to equi-biaxial tension. The Hessian matrix is computed by using the server mentioned above.
The mechanical properties of DNA are also critical, since in the cell DNA twists, bends, and packs into a small volume, which induce high stress and strain energy. In fact, the interchange between nutritional energy and strain energy of supercoiling is a mechanism for controlling growth in some organisms. The torsional properties of the double helix are also important for biological sequences. While NMA and ENM are in general applicable to DNA (and RNA), atomic modeling of DNA can still be quite time-consuming due to the large number of atoms involved. Thanks to the ultrahigh length-to-section ratio (up to 10,000,000), the DNA may be smeared out to behalve like a continous elastic rod with specified bending and torsional stiffnesses. The rod may be regarded as rigid against axial and lateral shear loads. If the stiffnesses are invariant with respect to the DNA sequence and local environment, in some cases closed-form analytical solutions are available based on the classical Kirchhoff elastic rod model.
Since both the magintude and directionality of the local stiffness strongly depend on the DNA sequence, the stiffness of the rod should vary in a prescribed manner, and the intrinsic curvature and twist also need to be incorporated into the approach. Moreover, the bending properties of the DNA are strongly affected by its immediate ionic environment. The solvent molecules can perturb the effective energy of the biomolecule at equilibrium through electrostatic interaction; moreover, the solvent molecules bombard the DNA and therefore cause an effective dragging force that influence the dynamics. The DNA also interact strongly and continuously with proteins, where the proteins could induce a particular trajectory to the axis of the double helix, to direct the bending/twisting/supercoil/folding/loop deformation so as to pack the DNA into a small volume.
While it is difficult to incorporate all these considerations into one analytical model, we note that the groups of L. Mahadevan and Klaus Schulten have made an important step forward, in the following paper
In this paper, they have extended the classical Kirchhoff model by incorporating the sequence-dependent intrinsic twist and curvatures, anisotropic bending stiffness, electrostatic interactions on DNA loops, and overdamped Brownian motion in a solvent via a Monte Carlo simulation. The deformed elastic rod is a scaffold on which all-atom structure of the entire DNA can be established, and the interaction between protein atoms and the atomic details recovered from the DNA rod model serves as the basis for a multiscale approach:
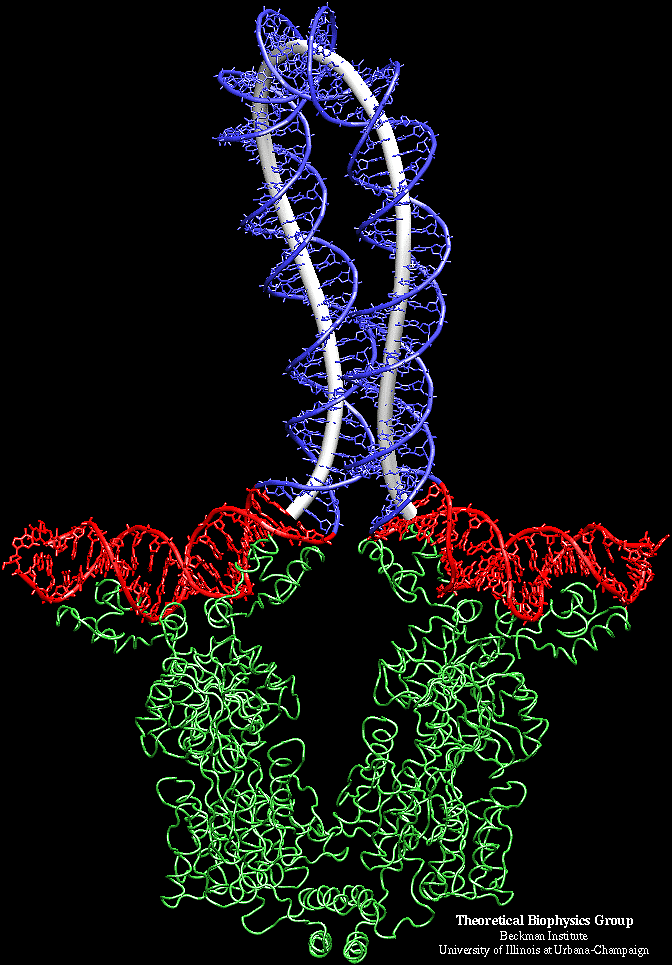
From a model system of lac repressor protein -DNA complex (the third figure on the left side is taken from Professor Schulten's website), the molecular dynamics (MD) simulation of the protein is coupled with the continuum DNA rod model through the "interface" at the beginning and ending of the loop. The MD simulation is carried out on the proteins, the end segments of DNA, and solvent. After every 10,000 time steps in MD simulation, the elastic rod model is called and DNA configuration is updated. The reaction forces at the end of DNA are then substituted into the MD calculation region and applied on relevant atoms. This multiscale approach, with certain parts of the complex treated by continuum "coarse grain" model, may find important applications for other macromolecule chains, as well as for studying the mechanochemical interactions among various biomolecules and systems.
{kind=link}
The analytical models of proteins and DNA introduced above are extensions of the classical mechanics theory. The initial points for discussing these papers are their basic assumptions, advantages and disadvantages, as well as how to improve and extend these models. Please feel free to initiate a "brainstorming" discussion on how mechanics may be applied to biomolecules and systems while remain failthful to the structural details.
temperature effects in DNA, RNA and proteins
Dear Xi,
I finished reading the papers you suggested. Thanks for providing very good references for this interesting topic. I would like to say something, although I know nothing about biomechanics.
How to analyze the temperature effects for DNA, RNA and proteins? How was the temperature of DNA, RNA, or proteins, calculated in the previous molecular dynamics analyses?
As discussed before (http://imechanica.org/node/1064), temperature, as well as other thermodynamic concepts such as entropy and the 2nd law of thermodynamics, will become confusing at the molecular scale.
The challenge is that, the temperature fluctuation will affect greatly the functioning of DNA, RNA, and proteins. It is a very important issue that we just cannot ignore.
In reply to temperature effects in DNA, RNA and proteins by Henry Tan
The biomolecules cannot be placed in an isolated system
Thank you Honglai. All components discussed here must be embedded within a cell and its surrounding environment and thus it is usually assumed the system temperature is a constant. In MD simulation that would be to put the entire system in a "water bath" with a constant temperature. The thermal fluctuation can, of course, influence the system equilibrium and dynamics (especially those from the solvent molecules). This is discussed in Mahadevan and Schulten's work.
In reply to The biomolecules cannot be placed in an isolated system by Xi Chen
how many atoms in a cell?
Xi,
how many atoms in a cell? Counted in molecules, what is the number?
In reply to how many atoms in a cell? by Henry Tan
over billions of molecules per cell
Honglai,
The actual number varies a lot because the cell size can be very different. On average a protein can contain millions of molecules, and a cell can easily contain over billions of molecules. See here for a rough estimation.
In reply to over billions of molecules per cell by Xi Chen
all-atom simulations
Thanks for the information.
I can understand now that all-atom simulations of the conformational changes of proteins can be prohibitively expensive.
In reply to all-atom simulations by Henry Tan
even more expensive for time scale
Since typical processes occuring in the cell, such as mechanotransduction, can take several ms, all-atom simulations are simply impossible without some biased tricks.
In reply to even more expensive for time scale by Xi Chen
MD Simulations
I think that the ultimate test for dynamical processes in biomolecules is simulation of the folding process going from a random coil state to a native (folded) state. Several papers have shown that through MD simulations one can simulate the folding of small peptides with less than about 30 amino acids. But since most enzymes is typically much larger and that the folding process in general on the time scale of ms compared to only ns for large systems in MD we still have a long way to go.
In reply to even more expensive for time scale by Xi Chen
time scale in cells
Time scales for physiological processes in cells can take more than ms, and in many cases much closer to s. There's then yet another huge leap towards thinking about biology and disease -- in humans we have to try and understand how processes taking place in cells on second-type time scales relate to diseases that appear in years. A similar problem exists in length-scales: a cell is tens of microns; a person is (usually!) several meters in size.
I applaud the advances in biomechanics at molecular length- and time-scales but hope that these advances are kept in the perspective of the larger picture. To actually use our understanding of molecular process to affect healthcare there's a large leap in scale, and this leap is not irrelevant in considering healthcare as at least one good reason for studying biomechanics (aside from basic understanding of the universe and the meaning of life, of course!)
In reply to time scale in cells by MichelleLOyen
You are right
Michelle, you are absolutely right, processes in cell can take a longer time. I referred to mechanotransduction (which is more mechanics-related and closely related with protein channels discussed in the jClub) to illustrate that, even a ms (or even μs) process is too difficult to be followed by all-atom simulations. Like you said, length and time scale challenges present in almost every material mechanics problems, not just biomechanics.
In reply to time scale in cells by MichelleLOyen
time scales in life
Well I think that you have a very good point there and I also often think that science has a problem with scales. We look at something small and want to understand the bigger picture. Sometimes it is fruitful but often it leads to "wrong" conclusions. But there is really no alternative way to do science because if we dont understand the basics we can not understand the advanced......
In reply to over billions of molecules per cell by Xi Chen
This is what I presumed.
This is what I presumed.
further model reductions on protein structures
Probably, it may be good to include Bahar's paper on protein modeling. First, we may consider her paper published in J. Mol. Biol. in 2003: Xu, C., Tobi, D., and Bahar, I., J. Mol. Biol., 333, 153-168 (2003). In her work, it was shown that conformation transition from T form (tense conformation: ligand-unbound state) to R form (relaxed conformation: ligand-bound state) was induced by purely elastic forces (entropic forces). That is, the conformational transitions are related to low-frequency normal modes responsible for entropic forces (thermal fluctuations). For understanding conformational transition by using NMA, one may also consider the following papers:
Zheng, W., Brooks, B.R., Biophys. J., 88, 3109-3117 (2005)
Ikeguchi, M., Ueno, J., Sato, M., Kidera, A., Phys. Rev. Lett., 078102 (2005)
Moreover, in recent year, there was an issue on further coarse-graining on protein structures. In Bahar's recent work, it was shown that collective motion of large proteins can be represented by small number of degrees of freedom. Specifically, in her work, the structure (~10^4 residues) of GroEL-GroES complex can be represented by only 30 nodal points. For details, you may refer to her paper: Chennubbhotla, C., and Bahar, I., Lecture Notes in Computer Science, 3909, 379-393 (2006). Further, I also provided a method of coarse-graining of protein structures. For details, you may look at the paper: Eom, K., Baek, S.-C., Ahn, J.-H., Na, S., J. Comput. Chem., in press, doi: 10.1002/jcc.20672.
In reply to further model reductions on protein structures by Kilho Eom
Thank you Kilho
Since the journal club could only include 3-5 papers for discussion, I only chose the most representative (and more pioneering) papers in this area (based on my humble and limited knowledge). I am aware of the three NMA/ENM papers you mentioned, which are more or less follow-ups on this topic. The papers you listed will be important for the readers to further understand the topic.
For coarse graining of proteins though, I feel it is more computational than analytical; plus, comparing with coarse graining of lipid, the coarse graining of protein is still at its infant stage and there are still many unsolved problems. Moreover our group is also doing research in this area. Overall I leave this topic out of this issue of journal club but I welcome discussion on this topic; or you may consider to include that in the theme of a future issue of journal club.
In reply to Thank you Kilho by Xi Chen
RE: unsolved problems on coarse-graining of proteins?
Hi, Chen. I am very thankful to your reply to my comments. You said that coarse-graining of proteins is still at infant stage and there are still many unsolved problems. As far as I guess, the coarse-graining of proteins is attributed to the fact that the native topology (represented by contact map - map indicates the native contact) plays a role in protein dynamics. That is, in general, the stiffness matrix for proteins based on native contacts becomes sparse matrix that leads to possibility of coarse-graining. I think that that is why further model reduction from Tirion's coarse-grained model is possible.
I have simple question for you: You said that "there are many unsolved problems". Can you give some example on your comment? To my best knowledge, the challenging problem is to understand protein dynamics and/or mechanics for large protein complexes that I am still working on. Especially, I am working on protein dynamics of large protein complex (e.g. GroEL-GroES) by using various model reduction methods. Also, I am working on coarse-grained model for mechanics of protein crystals.
Anyway, I would like to hear from you about some examples of unsolved problems in coarse-graining of proteins, and your opinion for further directions on protein modelings. Thank you again for your reply.
In reply to RE: unsolved problems on coarse-graining of proteins? by Kilho Eom
being more quantitative
While most coarse-grained models of proteins can give reliable information about directionality of motion (such as structural transition), but not about the magnitude of motion. It's usually straightforward to correlate known structural transitions to a set of normal modes (which are collective basis vectors). It's much more difficult to predict structural transitions based on a single structure; e.g., what happens after an ATPase binds ATP. In other words, the work that you mentioned is quite nice but not yet predictive. For more quantitative coarse-graining attempts, you may want to refer to Greg Voth's work.
how far do you plan to go in biomechanics?
Xi,
So, how far do you plan to go in biomechanics? Any particular research directions?
In reply to how far do you plan to go in biomechanics? by Henry Tan
biomechanics is one of our research areas
Honglai,
We are interested in mechanics of biomolecules in general but more focused on simulations (instead of modeling in this jClub article). Specifically, we are looking at mechanosensitive channels at this moment and developing multiscale simulation protocols. This will be one of our main directions in the coming years. Of course, just like you, we are also and equally interested in materials for energy, nanomechanics, thin films, nanoindentation, and solid-fluid interactions. I am updating my department webpage and should post some new information soon.