
Wei Gao
Department of Mechanical Engineering, University of Texas at San Antonio
In this journal club, we provide a brief summary on the concept, recent progress and tools of machine learning (ML) potential for atomistic materials modelling. We hope that it could benefit to the readers who are new to this filed and plan to develop their own or use others ML potentials. Comments and disscussions are welcomed.
1. Rising of Machine Learning (ML) Potentials
Atomistic simulation has become an important tool for mechanician to investigate the mechanical behavior of materials in a bottom-up approach. In the simulations, the interatomic potential that describes the interactions of atoms determines the fidelity of simulation results. The main disadvantage of the classical interatomic potentials is that they are limited by the fixed functional forms and small number of fitting parameters. As a result, they may not be able to provide reliable predictions, for example, in the mechanics problems under high stress, large deformation or close to failure. By contrast, ML potentials are not relying on a physical functional form (thus much more flexible), but must learn the physical shape of the energy surface from the training dataset. Therefore, if the training datasets (which usually come from first principle calculations) cover sufficient physics, a well-trained ML potential is able to provide accurate predictions that are comparable to first principle results. So far, we have seen many successful examples of ML potentials developed for various material systems such as [1-4].
Under the scheme of computation-guided materials design, combined with the development of big Data (such as Materials Project), ML potentials play important role to accelerate discoveries of new materials by quickly and accurately identifying material systems with properties of interest. For example, a recent study demonstrated that two novel ultra-incompressible hard materials MoWC2 and ReWB were identified (from a screening of 399,960 transition metal borides and carbides) and successfully synthesized from experiment [5]. In another example, the authors screened 132,600 compounds with elemental decorations of the ThCr2Si2 prototype crystal structure and identified a total of 97 new unique stable compounds, accelerating the computational time of the high-throughput search by a factor of 130 [6].
2. Types of ML potentials
ML potentials can be broadly split into two categories: (1) descriptor-based ML potential, in which the descriptors (also called “fingerprints”) are used to describe the environment of the atoms in a system, which is required to satisfy necessary rotational, translational, and permutational invariances as well as uniqueness [7], and (2) end-to-end ML potentials, which do not need fixed descriptors but instead learn atom environments directly from atom types and positions. Although the end-to-end ML potentials utilize more recent and advanced feature learning AI technology, there is still no evidence to show that end-to-end ML potentials outperform the descriptor-based ML potentials in terms of prediction accuracy.
3. Descriptor-based ML potentials
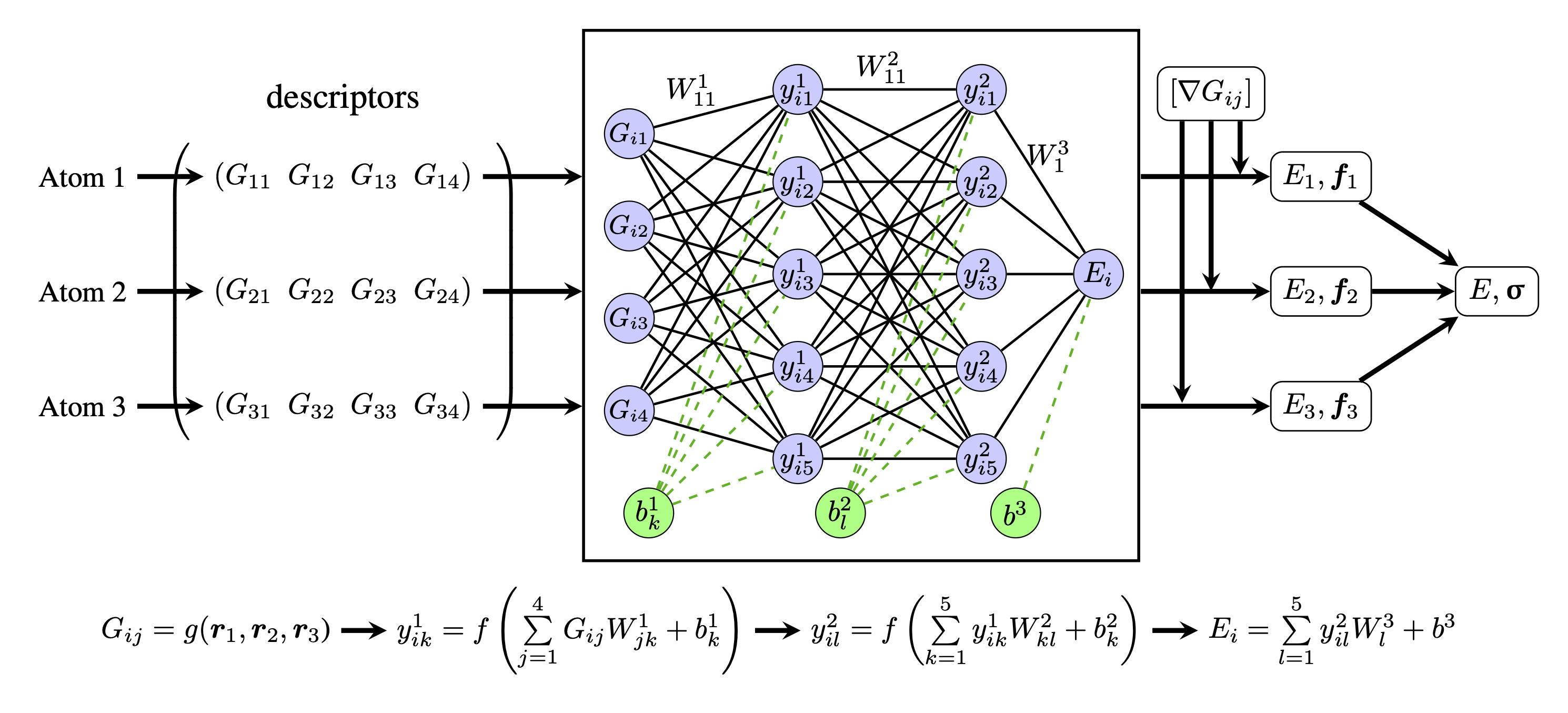
The descriptor-based ML potentials can be categorized by different types of descriptors and different types of ML models. We first present an elementary explanation on the working principle of descriptor-based potential, which is described in Fig. 1 using a neural network as the exemplary ML model. Consider a single element system containing 3 atoms. A descriptor is chosen to map the coordinates of each atom to a feature vector with 4 components (Gij). Then, the vectors are taken as inputs to the same neural network, which is composed by 2 fully connected dense layers (each with 5 neurons) followed by a linear layer at the end. The neural network can be considered as a high dimensional function that is parametrized with weight matrices and bias vectors (which are optimized during training). The equations in Fig.1 demonstrates the mathematical process inside the neural network, where W1, W2 and W3 are the weight matrices of each layer, b1, b2 and b3 are the corresponding biases, and f is the activation function of dense layer. The direct outputs of the neural network are the potential energies of the atoms. In addition, the atomic forces can be calculated by using the gradients of the descriptors (which have to be generated before training). The neural network can be trained by minimizing a loss function (e.g. mean average error) that can be measured by using total potential energy, atomic forces and stress. Once the neural network is trained, it can be used to predict per-atom energy and forces as well as stress of a new atomic system which may have different number of atoms but have to be encoded by the same type of descriptors.
Fig 1. A diagram of descriptor-based neural network potential for a single element system with 3 atoms.
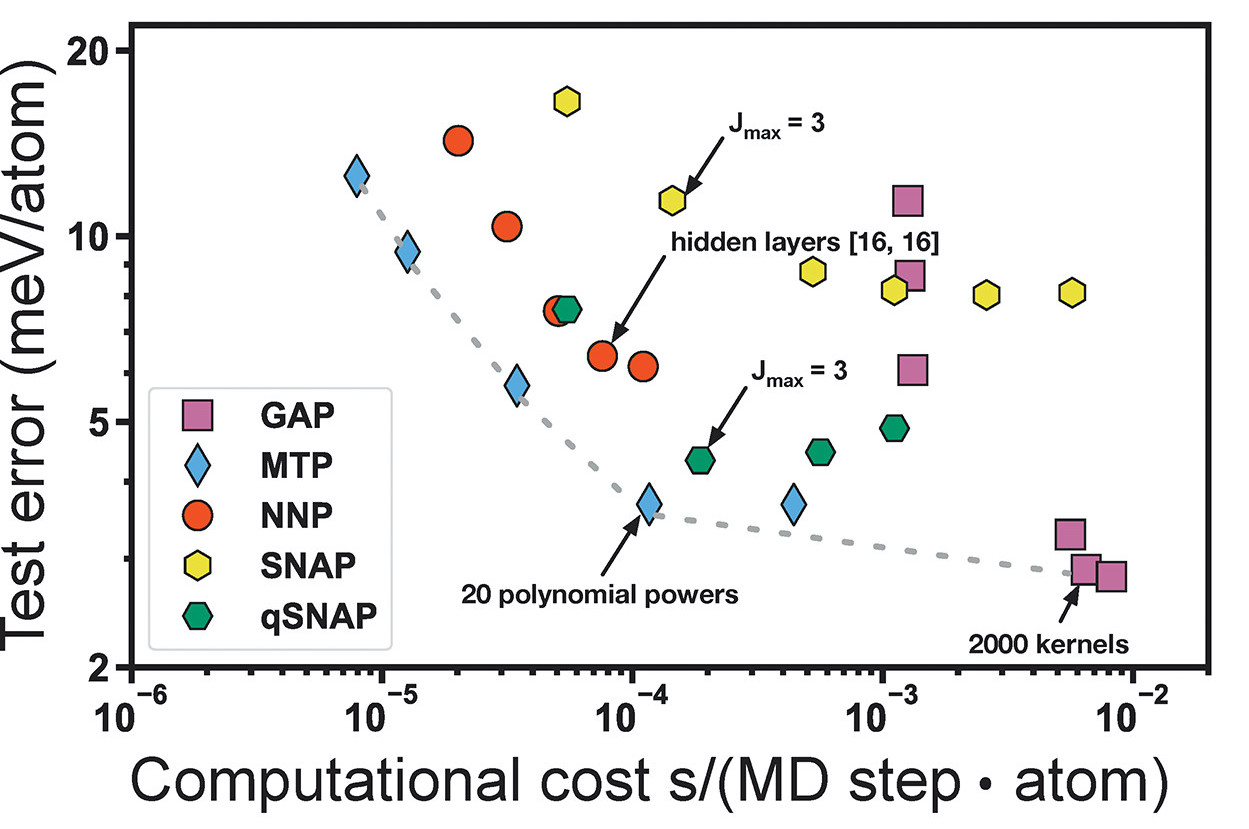
The framework of ML potential shown in Fig.1 can be customized by using different types of descriptors and different ML models, leading to many different types of descriptor-based ML potentials. The representative ones include: (1) Behler and Parrinello Neural Network potential (NNP) [8,9]: it uses atom centered symmetry functions (ACSF) as the descriptor and high dimensional neural network as the ML model, where ACSF describes atom local environment via a combination of radial and angular distribution functions; (2) Gaussian approximation potential (GAP) [10]: it uses Smooth Overlap of Atomic Positions (SOAP) as the descriptor and the Gaussian process regression as the ML model, where SOAP is a function expansion of local atomic density; (3) Spectral Neighbor Analysis Potential (SNAP) [11], which employs the bispectrum components (another type of function expansion of local atomic density) as the descriptor and uses a linear regression model to fit the data. (4) Moment Tensor Potential (MTP) [12]: it uses rotationally covariant tensors to describe the atomic local environment along with a linear regression model, where the tensors can be treated as a series of radial and distribution functions like ACSF. Apparently, the performance of a ML potential depends on both the choice of descriptor and ML model. A recent study [13] compared the performance of descriptor-based ML potentials on a range of crystal structures with a single CPU core, as shown in Fig. 2, which shows the “optimal” MTP, NNP and SNAP models tend to be 2 orders of magnitude less computationally expensive than the “optimal” GAP model, and better accuracy can only be attained at the price of greater computational cost. At the end, there is no absolute winner among these methods. The performance may be improved by mixing the descriptors and ML models although we have not seen such kind of study yet.
Fig 2. Performance comparison of several descriptor-based ML potentials on a range of crystal structures [13].
4. End-to-end graph-based ML potentials
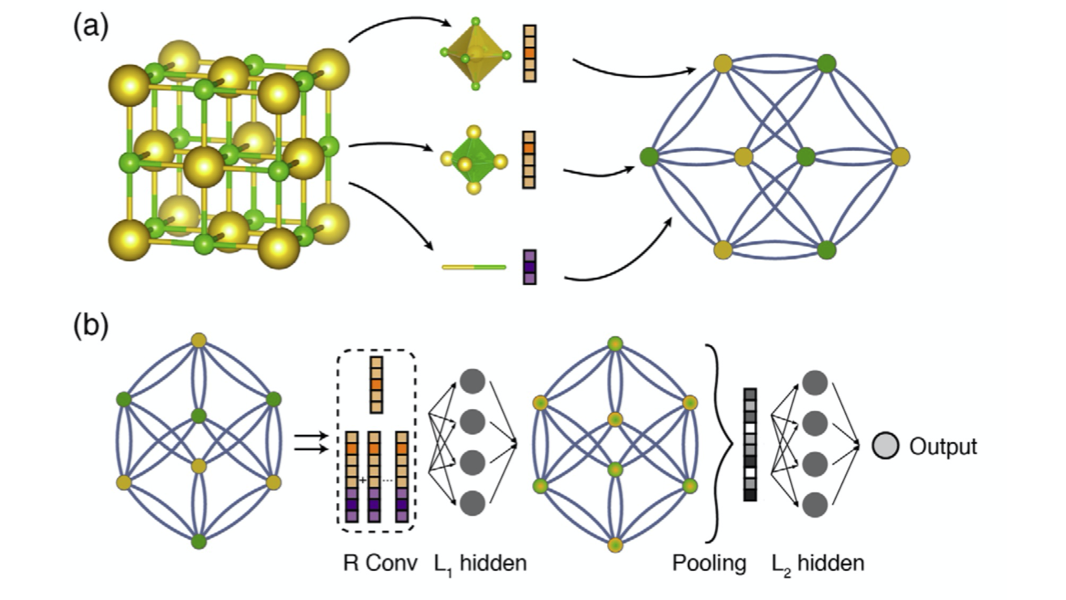
The end-to-end ML potentials are all based on the concept of using graph to describe atomic system, in which the topology of the ML network is based on the topology of atomic structures. The early development (from 2015 to 2017) of end-to-end ML potentials are mostly focus on molecules. In 2017, a group of Google research scientists abstracted the commonalities among the existing end-to-end graph-based potentials in literature using a Message Passing Neural Networks (MPNNs) framework [14], in which feature vectors are defined on the atoms and bonds that are respectively represented as nodes and edges. The message is simply a function acting on the feature vectors. Such message function is passed iteratively among the nodes, in order to learn the graph structure of the atomic system and update the feature vectors. Such iterative process is similar to that used in convolutional neural networks, so it can be called convolutions. It is noted that using different message function can lead to a variety of graph-based ML potentials. More recently, upgraded methods have been applied to solid-state materials. Here, we introduce three representative ones. (1) Crystal Graph Convolutional Neural Networks (CGCNN) [15], which can be viewed as a special case of MPNNs with a special message function. Fig. 3 shows the schematics of a crystal graph in which the atoms and bonds become nodes and the edges. The neural network is composed by two parts: in the first part, feature vectors are learned through message-passing R convolutions followed by L1 fully connected dense layer; in the second part, the feature vectors are pooled into a single feature vector which is then passed to another L2 fully connected dense layer for computing outputs. Apparently, the feature learning is the unique part as compared to the neural network shown in Fig. 1. (2) SchNet [16], which is a deep neural network architecture based on continuous-filter convolutions. The continuous-filter, as a generation of the classical filter used for evenly spaced data such as image pixels, was introduced to handle unevenly spaced data (atoms positions). (3) MatErials Graph Network (MEGNet) [17], which is based on a more recent graph network and can be viewed as a superset of previous graph-based neural networks. One unique feature in MEGNet is that it incorporates global state variables (e.g. temperature) into training in order to directly predict state-dependent properties such as free energy. It is noted that SchNet and MEGNet can be applied to both molecules and solid-state materials.
Fig. 3. Illustration of the crystal graph convolutional neural networks [15]. (a) Construction of the graph. (b) Structure of the neural network including the layers for feature learning (R convolutional layers and L1 hidden layer) and the layer for computing outputs (L2 hidden layers and output layer).
5. Tools for ML potentials
In recent years, there have been many ready-to-use open source packages developed along with the development of various new ML methods for both descriptor-based and end-to-end ML potentials. Even for the same type of method, different tools have been developed to achieve some special needs. Here, we introduce several well-documented and maintained tools that have been connected to well-established atomistic simulation packages such as LAMMPS and ASE. (1) n2p2: it is a package written in C++ based on the Behler-Parinello neural network potentials. It can be used to train the potential with potential energy and atomic forces and provides an interface to LAMMPS. This tool coded all the aspects of ML potential together including feature generation, neural network training and prediction. This makes it friendly to users who can simply treat it as a Blackbox. However, it is also more challenging to add some new features, such as adding stress as one of the training variables. (2) GAP: a Fortune package that is wrapped with a Python interface, developed by the authors who proposed GAP method introduced in section 3. It runs inside the QUIP program, which is a plugin package in LAMMSP, so GAP is connected to LAMMPS through QUIP. In addition, it is also connected with ASE. (3) SchNetPack: a python based package that includes the end-to-end ML potential SchNet introduced in section 3, where the ML models are implemented through Pytorch. The package is connected to ASE but not to LAMMPS. (4) KLIFF: a more recent package developed to facilitate the entire interatomic potential development process. The package can be used to fit both traditional empirical potentials as well as Behler-Parinello type of neural network potentials (implemented with PyTorch). It is integrated with KIM package that is maintained in the same group and able to communicate with many materials simulation packages including LAMMPS and ASE. (5) AtomDNN: a package recently developed in our group for training Behler-Parinello type of neural network-based potentials, where we use LAMMPS as a Calculator to generate descriptors and use tf.module in Tensorflow2 to train the potential with energy, forces and stress. The trained potential is integrated with LAMMPS using Tensorflow C APIs that are directly compiled with LAMMPS. The code is user friendly and can be easily customized to achieve special needs.
6. Summary and Outlook
ML potentials have already demonstrated their significance for atomistic simulations, as well as for the design of new materials through efficient property predictions. The field, especially the end-to-end graph-based potentials, is still under rapid development along with the development of new AI technologies. For mechanicians, ML potentials provide a new way to train the potentials with meaningful dataset that contains rich mechanics information of interest, so that the target mechanics problems can be studied at atomic scale with high fidelity. Machine learning has also been applied to many other mechanics problems in the meso and continuum scale to improve the existing mechanics or materials models or learn new models from data. It may be possible in the future to leverage the data from multiple scales to train meaningful multiscale material models that can predict comprehensive macroscopic materials behavior based on atomistic and meso scale structures.
Reference:
[1] Bartók, Albert P., et al. "Machine learning a general-purpose interatomic potential for silicon." Physical Review X 8.4 (2018): 041048.
[2] Rowe, Patrick, et al. "Development of a machine learning potential for graphene." Physical Review B 97.5 (2018): 054303.
[3] Wen, Mingjian, and Ellad B. Tadmor. "Hybrid neural network potential for multilayer graphene." Physical Review B 100.19 (2019): 195419.
[4] Jain, Abhinav CP, et al. "Machine learning for metallurgy III: A neural network potential for Al-Mg-Si." Physical Review Materials 5.5 (2021): 053805.
[5] Zuo, Yunxing, et al. "Accelerating Materials Discovery with Bayesian Optimization and Graph Deep Learning." arXiv preprint arXiv:2104.10242 (2021)
[6] Park, Cheol Woo, and Chris Wolverton. "Developing an improved crystal graph convolutional neural network framework for accelerated materials discovery." Physical Review Materials 4.6 (2020): 063801.
[7] Bartók, Albert P., Risi Kondor, and Gábor Csányi. "On representing chemical environments." Physical Review B 87.18 (2013):184115.
[8] Behler, Jörg, and Michele Parrinello. "Generalized neural-network representation of high-dimensional potential-energy surfaces." Physical review letters 98.14 (2007): 146401.
[9] Behler, Jörg. "Atom-centered symmetry functions for constructing high-dimensional neural network potentials." The Journal of chemical physics 134.7 (2011): 074106.
[10] Bartók, Albert P., et al. "Gaussian approximation potentials: The accuracy of quantum mechanics, without the electrons." Physical review letters 104.13 (2010): 136403.
[11] Thompson, Aidan P., et al. "Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials." Journal of Computational Physics 285 (2015): 316-330.
[12] Shapeev, Alexander V. "Moment tensor potentials: A class of systematically improvable interatomic potentials." Multiscale Modeling & Simulation 14.3 (2016): 1153-1173.
[13] Zuo, Yunxing, et al. "Performance and cost assessment of machine learning interatomic potentials." The Journal of Physical Chemistry A 124.4 (2020): 731-745.
[14] Gilmer, Justin, et al. "Neural message passing for quantum chemistry." International conference on machine learning. PMLR, 2017.
[15] Xie, Tian, and Jeffrey C. Grossman. "Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties." Physical review letters 120.14 (2018): 145301.
[16] Schütt, Kristof T., et al. "Schnet–a deep learning architecture for molecules and materials." The Journal of Chemical Physics 148.24 (2018): 241722.
[17] Chen, Chi, et al. "Graph networks as a universal machine learning framework for molecules and crystals." Chemistry of Materials 31.9 (2019): 35
| Attachment | Size |
|---|---|
| NNP.png | 553.58 KB |
| comparison.jpeg | 150.71 KB |
| CGCNN.png | 441.73 KB |
{kind=link}
{kind=link}
{kind=link}
Hi Wei, thanks so much for
Hi Wei, thanks so much for offering such an informative tutorial. Just one quick question: For descriptor-based ML potentials, can we directly use the coordinates of atoms as the input for the neural network? If we have to convert the coordinates into descriptors as the inputs, what is the standard procedure to do so? Many thanks!
In reply to Hi Wei, thanks so much for by Zheng Jia
Hi Zheng,
Hi Zheng,
Thanks for your intetests! Most of the descriptor-based packages, as listed in section 5, only ask for atom coordinates as inputs, which are transformed to descritpors internally through descritpor functions (such as ACSF, SOAP, as listed in section 3). Some packages (such as n2p2) implicitly run descritpor generation so that it's not convenient (or impossible) for users to see the descriptors. In our code (AtomDNN), we compute the descritpors (ACSF and SOAP are currently supported) through customized LAMMPS Compute commands (can be found here with an example) and make the descriptor generation explict through a well defined data pipeline. In this way, descritpors can be computed without training the network. There are other tools, such as DScribe, which is only used for computing many types of descriptors. However, we found it is slow for computing the derivatives of descriptors (which are needed for compute atomic force and stress). For SOAP descriptors, you can also use QUIP to get the descriptors directly.
Wei
very nice!
Dear Wei,
Thank you for this nice summary on machine learning potentials. I enjoyed reading it and learning the state of the art. I have two questions in mind:
(1) How to train a machine learning model? As you noted, the performance of a ML potential depends on both the choice of descriptor and ML model. In addition, I think it also depends on the training and the data used for training. Can you elaborate on the steps taken to train a ML model?
(2) How to use one of the ML tools (Section 5) as a blackbox for atomistic simulations? I assume that these tools have been trained one way or another and thus are ready to be used directly in place of the standard empirical potentials (e.g. in LAMMPS). Is it as simple as that?
Again, I am impressed by how much you have done in this area, and congratulations on your recent CAREER award!
Rui
Dear Rui,
Thank you so much for the kind and encouraging words. To your questions:
(1) You made an important point: the performance of ML potential is highly dependent on the training data. Only high quality data that covers the essential physics of interests can produce reliable ML potential. Therefore, developing ML potential starts from the data generation, usually by using DFT calculations. First, a variety of atomic structures have to be carefully prepared. For example, the atomic structures could come from the random perturbation of atom positions and lattice constants based on a perfect crystal structure. In addition, typical defect structures can be built into the data. Recently, we also use atomic structures coming from Nudged elastic band and Dimer calculations to inform the data with phase transition information and to better sample the potential energy surface. Nowadays, more and more researchers share materials dataset publicly, so that one could re-use those already available datasets and enrich them with specific physics of interests if needed. After atomic structures are determined, one just need to run DFT calculations to get the outputs of interests that will be used for training, such as energy, force, and stress.
The inputs to a ML model are the well-prepared atomic structures. These structures are converted to descriptors when one uses a descriptor-based methods. This conversion can be done automatically within the packages described in section 5 without interference. The outputs to a ML model depend on the application. Most of the time, the potential energy, atomic force and stress are used as outputs. ML model (e.g. neural network) can be conveniently built using some machine learning platforms such as Tensorflow and Pytorch, which provide the library functions for training the model. There are many hyper parameters that can be tuned in order to achieve a good convergence when the loss settles to within an error range. After training is done, the ML model can be saved as the ML potential, which can be used later like classical potentials.
(2) The tools described in section 5 can be used as black box, and the products (ML potentials) can be directly used for MD or MS simulations. Those tools except Schnet can be all connected to LAMMPS and used just like classical potentials.
Wei
Impressive!
Dear Wei,
Thanks for a very informative edition of the journal club.
Though I have been in the ML field for some time, when it comes to this particular area (ML Potentials for Atomistic Simulations) I am an absolute newbie. In fact, this journal club edition is my first ever contact with this research area. ... It does look like there has been a great deal of activity in this area in the recent times, and people seem to have approached the problems with a lot of creativity too. All in all, very impressive! [Even if you set everything else aside, anything like "accelerating the computational time of the high-throughput search by a factor of 130" just has to be impressive!]
OK, now, allow me a couple of newbie questions...
How do these approaches work for systems / phenomena involving polarization? Any work or notable results in this direction?
How precisely does the gain in the high-throughput search come about? ... If I understand it right, these potentials are just going to get incorporated into the MD / atomistic packages like LAMMPS, right? So, speaking simple-mindedly, the run-time computational complexity should also stay more or less the same, right? If so, how come there still is a gain?
Best,
--Ajit
In reply to Impressive! by Ajit R. Jadhav
Hi Ajit,
Hi Ajit,
Thanks for your interests and questions. To your first question, yes, the polarization effect could be described by ML potential as long as the potential is trained with the information of charges. There is a recent work (Nature communications 12.1 (2021): 1-11) specifically targeting this type of problem. Your second question is about the gain of ML potential. The main advantage of ML potentials as compared to classical potentials is that they can be much more accurate, although they are still generally slower than classical potentials.
Wei Gao
Thanks for the excellent review!
Dear Wei,
Thank you for summarizing the recent developments in ML potentials for MD simulations. I'm not working in this specific field. Reading your review satisfies lots of my curiosities.
I have 2 questions to ask here.
(1) In MD simulations, some interatomic potentials can capture bond breaking/formation, like Reaxff; some potentials cannot do so. And you mentioned that the ML potentials are learned from DFT. So are the existing ML potentials capable of capturing the bond breaking/formation? Are there any successful examples?
(2) You mentioned that the early development of end-to-end graph-based ML potentials was focused on molecules. Does it mean the end-to-end graph-based ML potentials are a better choice for polymer systems?
Thanks,
Haoran
In reply to Thanks for the excellent review! by Haoran Wang
Dear Haoran,
Dear Haoran,
Thanks for your interests and questions.
(1) Like those reactive potentials, there is no need to define fixed bonds in the ML potentials that are trained with DFT data, so ML potentials are able to capture bond breaking/formation. There are some good examples, such as the references [1-4] listed in the text.
(2) Molecules can be more conveniently descripted by a graph, so they are first studied with end-to-end ML potentials. However, descriptor-based ML potentials also have been used for molecules. The performance of the potentials is not only dependent on the ML model, but also many other factors, such as the quality of dataset, the choice of descriptors (or the quality of feature learning), as well as the rigor of training and validation process. Therefore, I have not seen a rigorous comparison between descriptor-based and end-to-end ML potentials in terms of prediction accuracy. If someone wants the machine to learn as much as possible from data (maybe better than human designed descriptors), then end-to-end model wins. This motivation is driving the development of new methods along with the rapid development of AI technology. However, at the moment, descriptor-based ML potentials are better connected to large scale simulator such as LAMMPS, so it may be a good choice to start with descriptor-based potentials if the final target is for large scale MD simulation.
Best,
Wei Gao
Timely topic!
Hi Wei,
Many thanks for this timely topic and for leading the great discussion.
Added to your summary, there are a few related works that might be useful:
1) The ML potential tool, such as the DeepMD is another excellent package, linked with LAMMPS to use. The DeepMD package can reproduce the temperature-pressure phase diagram of water molecules in the recent PRL paper, which is an intriguing demonstration for many mechanics problems under extreme environments. It also won 2020 ACM Gordon Bell Prize at SC20!
2) Physics-informed ML potential could be particularly useful since we want to avoid the unphysical interactions (energy, force, or stress) during the mechanical deformation. Imagining that we only train the ML potential from equilibrium conditions, it could not be used for large deformation simulations:) The recent physically informed artificial neural networks for atomistic modeling of materials can nicely address this issue, opening another avenue for ML potential to be applicable for large deformations, fractures, etc.
3) ML potential is not only useful for all-atom molecular simulations, but also for coarse-grained molecular simulations. When the ML potential is trained based on DFT data, it can reproduce the quantum accuracy, with efficiency as typical all-atom molecular simulations. Similarly, the ML coarse-grained model can archive the all-atom accuracy, with much less computational cost. It will open another door for us to model many large-scale mechanics problems with less computational time. We have a recent review article on this topic, Machine Learning of Coarse-Grained Models for Organic Molecules and Polymers: Progress, Opportunities, and Challenges.
Again, these are just my two cents.
I look forward to your fascinating works in this area and more discussions :)
Best, Ying
In reply to Timely topic! by Ying Li
Dear Ying,
Dear Ying,
Thank you for the informative comments! As you mentioned, DeepMD is certainly another useful tool. PINN is also an interesting direction which I consider as a combintation between classical empirical potential and ML potential. Thanks for sharing your review article on ML potential for CG systems.
Best,
Wei