
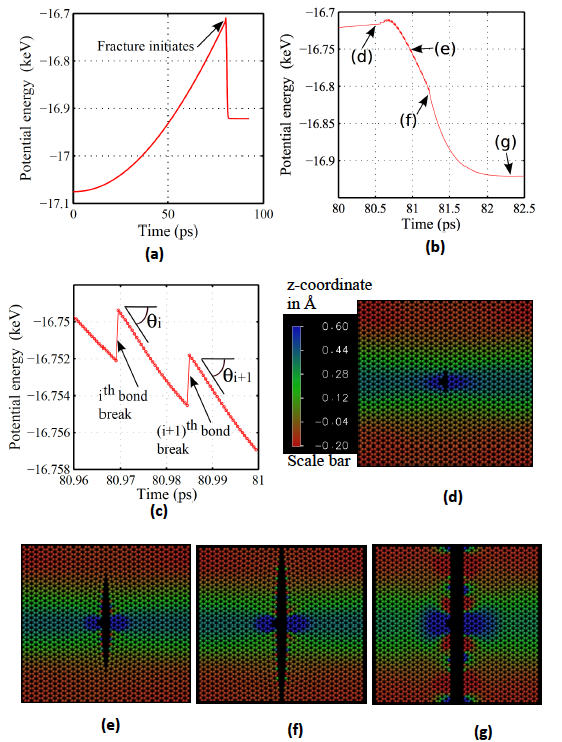
In our recent paper, atomistic and continuum modelling of temperature-dependent fracture of graphene, we directly calculate J integral using the data obtained from molecular dynamics simulations. Fig. 1 outlines the calculation procedure of JIC. Fig. 1(a) shows the changes in potential energy with time (or applied strain) in an armchair graphene sheet with a size of 7.6 nm × 7.6 nm. A crack, length of ~0.7 nm, is placed in the centre of the sheet. Periodic boundary conditions are used along in plane directions. Simulation temperature is 1 K, and strain rate is 0.001 ps-1. Fig 1(b) shows the variation in potential energy during the crack propagation. It can be seen that the potential energy increases just after the crack starts to propagate (around point d). This is due to the chemical potential energy release from carbon-carbon bond breaking overcomes the strain energy release by crack propagation. As more bonds break, the strain energy release due to crack propagation starts to govern the total strain energy release. The crack propagation at various stages (marked as d to g in Fig. 1(b)) is shown in Fig. 1(d) to Fig. 1(g). The figures show that the crack propagates symmetrically. The out of plane deformation of the sheet, as shown in the video 1, prevails.
Fig. 1 Calculation of energy release rate of graphene. (a) Variation of potential energy with time. (b) and (c) variation of potential energy during the crack propagation. (d) to (g) show the crack propagation in graphene. The corresponding positions of Fig. (d) to (g) in the potential energy-time curve are marked in Fig. (b)
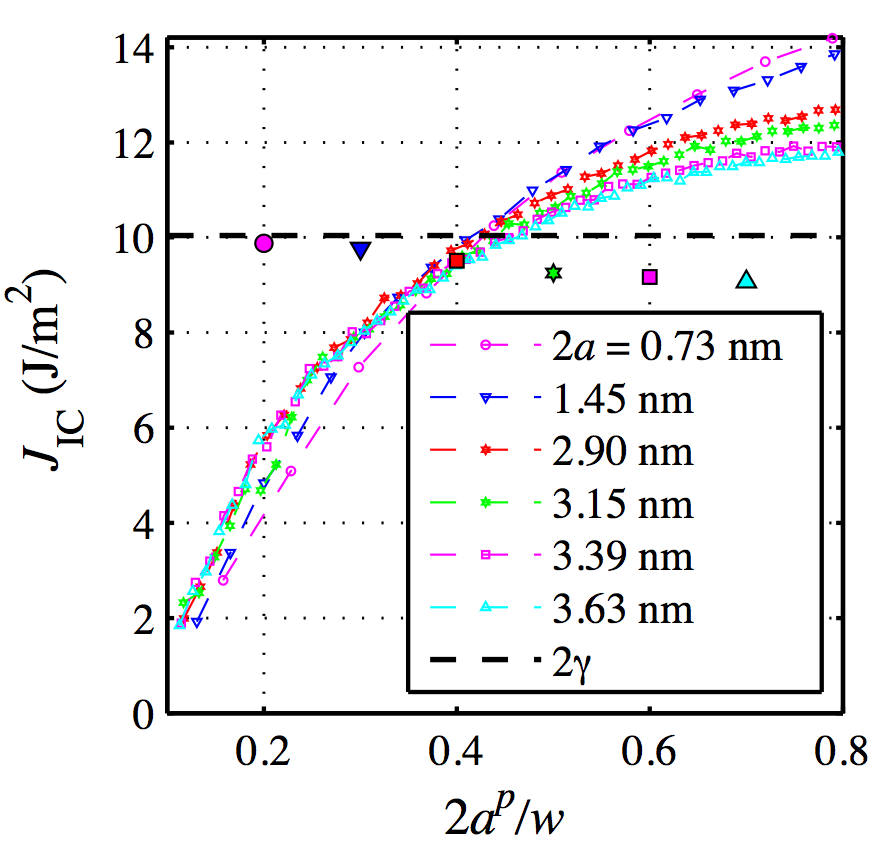
The slope of the piece wise continuous curves in Fig 1(c) is proportional to the critical value of J integral (JIC). Figure 2 shows the variation of JIC with the propagated crack length (2ap), which has been normalized with respect to the width of the sheet (w). The value of 2ap/w is approximately 0.1 when a crack starts to propagate since w is kept around 10 times the initial crack length (2a) to avoid the effects of finiteness. When 2ap/w reaches 1, the periodic cracks start to interact with each other. Therefore Fig. 2 shows the value of JIC up to 0.8 of 2ap/w, where the periodic cracks do not interact with each other for the smallest sheet considered (i.e. w = 7.6 nm).
Fig. 2 Variation of JIC of armchair graphene with propagated crack length (2ap) for various initial crack lengths (2a). The solid symbols indicate the average value of JIC (JIC,avg) at various crack lengths. The left most solid symbol is the JIC,avg for 2a = 0.73 nm and other marks are in ascending order of initial crack lengths. The right most symbol is the JIC,avg for 2a = 3.63 nm.
| Attachment | Size |
|---|---|
| change in j with crack propagation.png | 121.8 KB |
| J from MD.png | 345.93 KB |
{kind=link}
{kind=link}