
Journal Club for April 2019: Self-Healing Soft Materials: from Theoretical Modeling to Additive Manufacturing
Qiming Wang
University of Southern California
1. Introduction
Self-healing polymers have been revolutionizing the man-made engineering society via bringing in the autonomous intelligence that widely exists in Nature. Self-healing polymers have been applied to a wide range of engineering applications, including flexible electronics [1], energy storage [2], biomaterials [3], and robotics [4]. Motivated by these applications, various self-healing polymers have been synthesized during the past years [5-9]. They typically fall into two categories. The first category is “extrinsic self-healing” that harnesses encapsulates of curing agents [10,11]. The second category is “intrinsic self-healing” that harnesses dynamic bonds, such as dynamic covalent bonds [12-15], hydrogen bonds [16,17], and ionic bonds [18,19]. The dynamic bonds can autonomously reform after fractures or dissociations. This blog entry primarily focuses on the second category.
Despite the success in syntheses and applications, existing self-healing polymers are facing two critical bottlenecks. The first bottleneck is in the theoretical modeling of interfacial healing behavior [5]. Back in the 1980s, scaling models were proposed for the interpenetration of polymer melts [20,21]. After entering the 21st century, molecular dynamics simulations were employed to understand healing behaviors of polymers [22,23]. Although bulk healing [24,25] and high-temperature welding [26] have been modeled in recent years, how to analytically model the interfacial healing of self-healing polymers is still elusive. The missing of this theoretical understanding would significantly drag down the innovation of self-healing polymers to achieve optimal healing performance.
The second bottleneck is in the 3D-shaping method for self-healing polymers. A number of promising applications of self-healing polymers demand complex 2D/3D architectures, such as robotics [27], structural materials [28,29], architected electronics [30], and biomedical devices [3]. However, the architecture demand of self-healing polymers has not been sufficiently fulfilled, because existing 3D methods of shaping self-healing polymers are limited to molding [4,31] and direct-writing [32-34].
This blog entry outlines recent research efforts of Qiming Wang group at the University of Southern California in unblocking the above two bottlenecks: theoretical modeling [35-39] and additive manufacturing [40] of self-healing polymers. Challenges and opportunities are highlighted at the end of the blog.
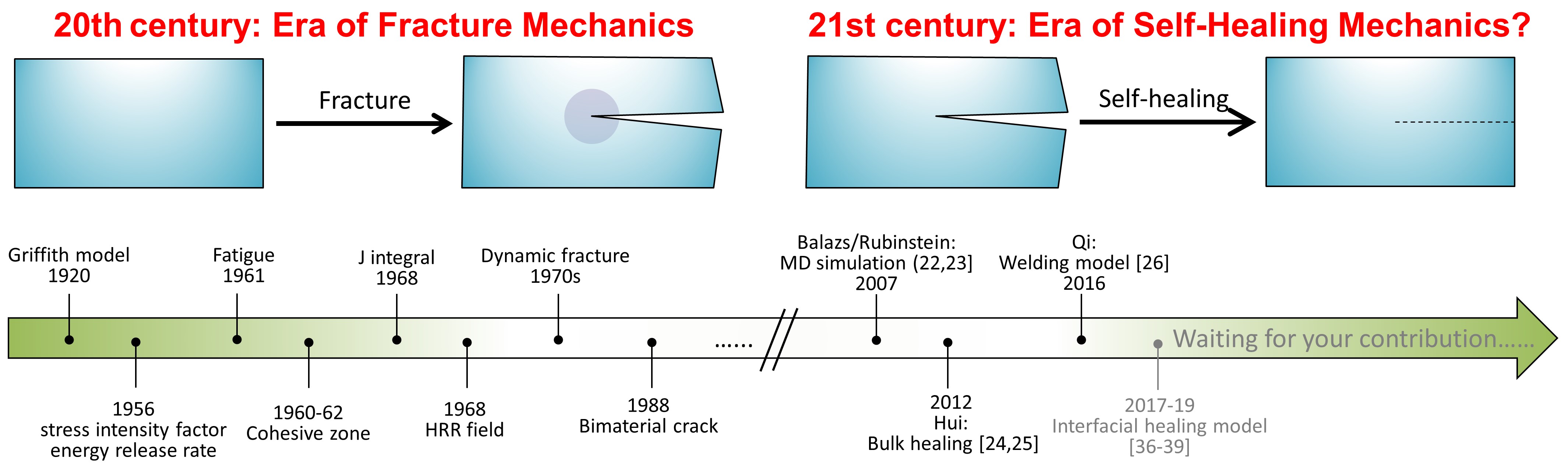
Jumping out of this blog, we would like to cast a vision on the field of solid mechanics shown in Fig. 1. Promoted by the US Navy after World War II, Fracture Mechanics enjoyed a golden era in the 20th century [41]. Question: several decades later when people look back, will they consider the 21st century to be the golden era of Self-Healing Mechanics? How do you think about the status/future of Self-Healing Mechanics? Please share your thoughts in the comment box.
Figure 1. Development of Fracture Mechanics and Self-Healing Mechanics.
2. Theoretical modeling of self-healing polymers
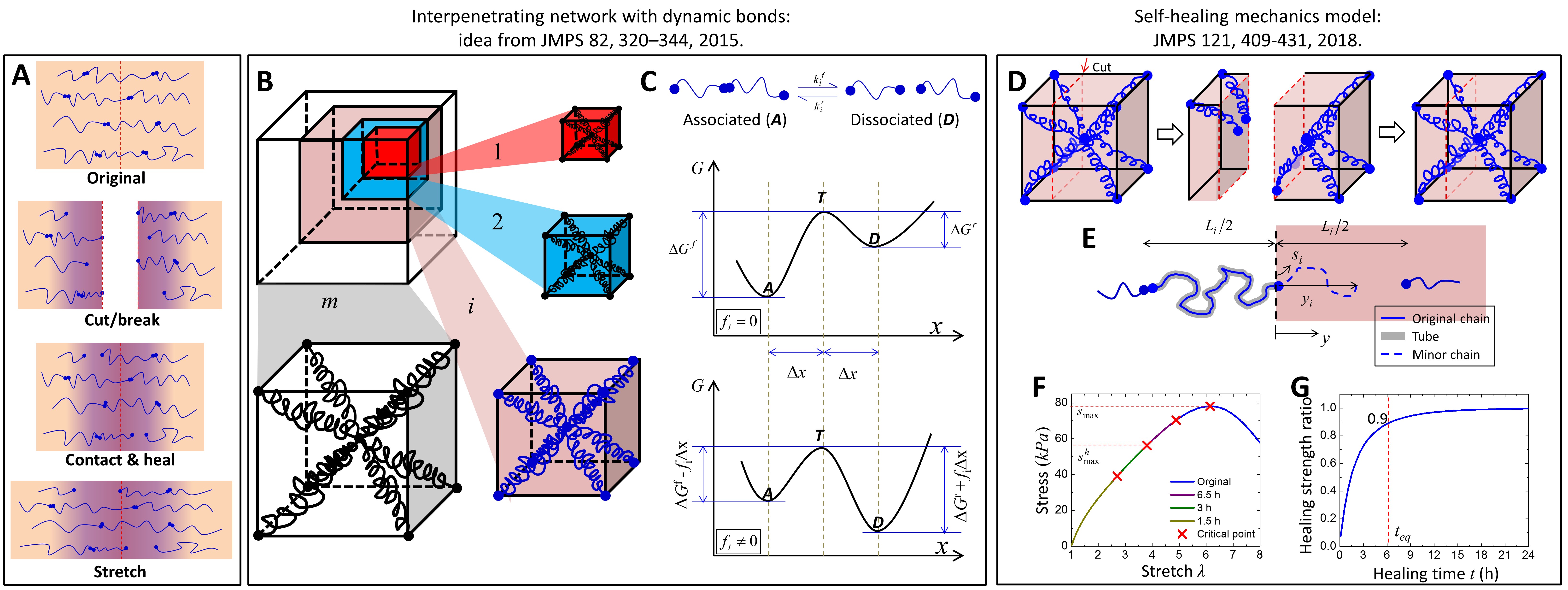
We consider the healing process of a polymer network linked by dynamic bonds shown in Fig. 2A. The polymer is cut into two parts and then contact back. After a period of healing time, the sample is stretched until rupture. The self-healed sample is composed of two segments (Fig. 2A): a small “self-healed segment” with re-bridged polymer chains (purple) and two “virgin segments” with intact polymer networks (light pink). The modeling effort is devoted to theoretically quantifying the relationship between the healing percentage and the healing time. The healing percentage is indicated by the ratio between tensile strengths of the healed and the original samples, because most of the researchers in the self-healing community use the tensile strength as the indicator [5-9].
Figure 2. (A) Healing process. (B) Interpenetration network model. (C) Bell-like model for a dynamic bond. (D) Conceptual self-healing model. (E) Diffusion and binding of a polymer chain. (F) Predicted stress-stretch behaviors of original and self-healed polymers. (G) Predicted relation between the healing strength ratio and healing time[37].
2.1. An interfacial self-healing model
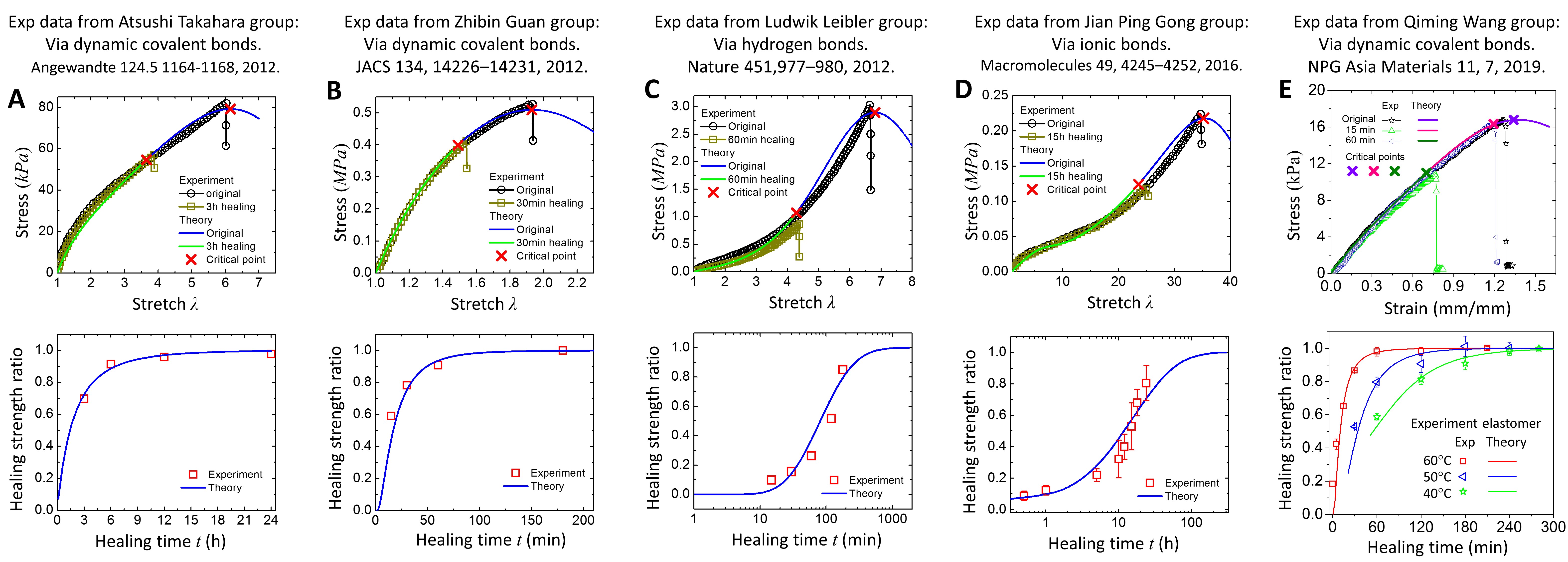
To theoretically model the interfacial healing of self-healing polymers crosslinked by dynamic bonds, we have two technical challenges: (1) how to understand the mechanics of dynamic-bond-linked polymer networks, and (2) how to understand the network evolution during the healing process. To address the first challenge, we employ an interpenetrating network model that many types of networks interpenetrate each other in the material space (Fig. 2B) [42]. Each type of network is composed of polymer chains of the same length and linked by dynamic bonds. The chain-lengths among different networks follow an inhomogeneous statistic distribution. Under stretch, dynamic bonds obey force-dependent chemical kinetics to transform between the associated state and the dissociated state (Fig. 2C). The force-dependent chemical kinetics can be described by a Bell-like model [43]. To address the second challenge, we consider the healing process as a coupled behavior of inter-diffusion of dissociated chains and re-binding of dissociated dynamic bonds (Fig. 2DE). The curvilinear motion of the polymer chain can be explained by a reptation-like model [44,45], and the binding kinetics by the Bell-like model [43]; therefore, the interpenetration of the polymer chain across the fracture interface can be modeled as a diffusion-reaction system [36,37]. After addressing the above two challenges, we can predict stress-strain behaviors of the original and the healed self-healing polymers (Fig. 2F). As the applied stretch increases, more and more dynamic bonds are dissociated, and the corresponding stress increases and then decreases. The maximal stress (tensile strength) is corresponding to the material rupture. With increasing healing time, the tensile strength of the healed polymer increases until reaching a plateau that is the tensile strength of the original polymer. In this way, we can predict the relation between the healing percentage (healed/original strength) and the healing time (Fig. 2G). Our theory can be used to explain self-healing behaviors of polymers crosslinked by various dynamic bonds, such as dynamic covalent bonds [14,15,40], hydrogen bonds [16], and ionic bonds [19,46] (Fig. 3).
Figure 3. Comparison between theoretical and experimental results of self-healing polymers [37].
2.2. Effect of polymer network architecture
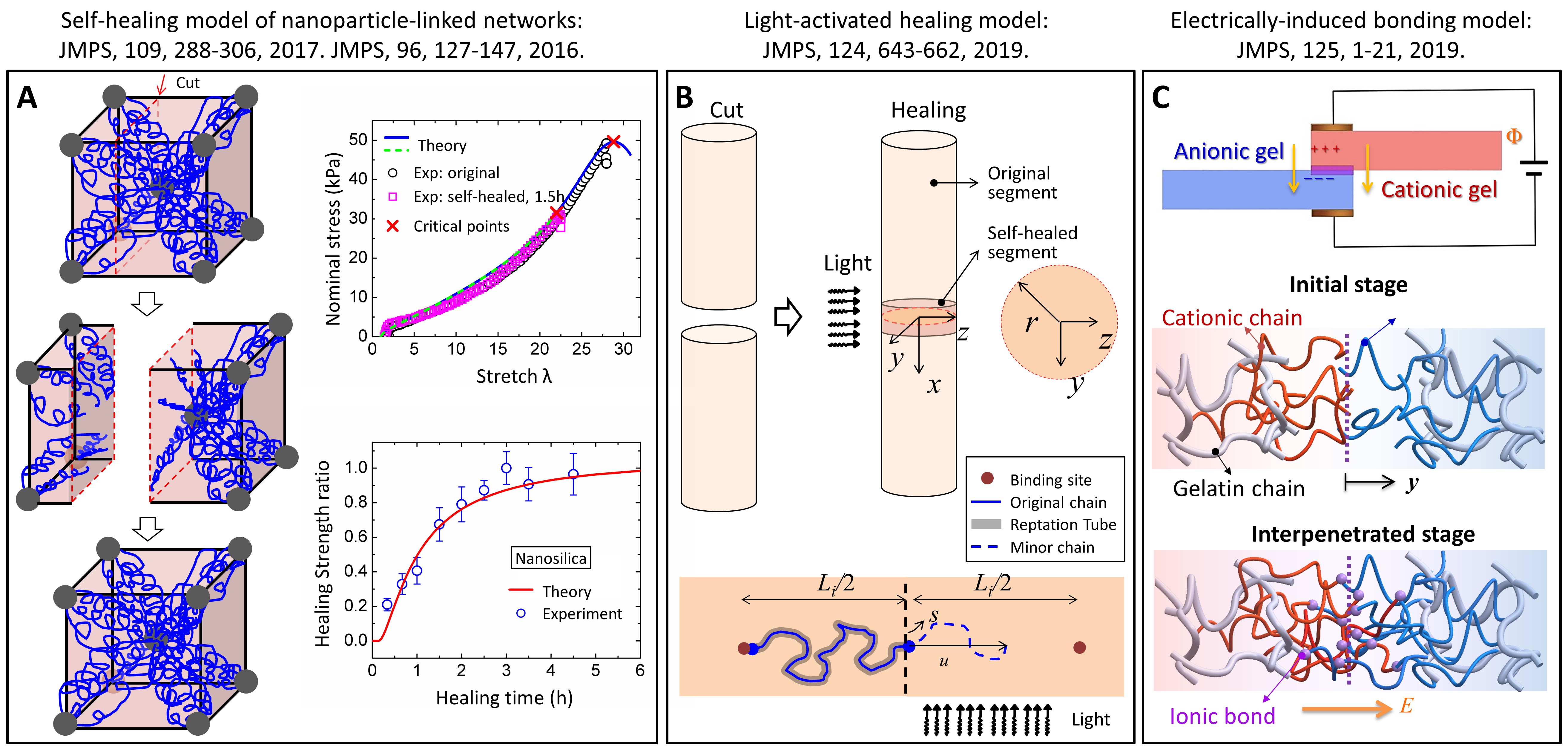
The self-healing model is expected to be extended to explain self-healing polymers with various network architectures [6,47]. Self-healable nanocomposite hydrogel is a good example: polymer chains are linked by multifunctional nanoparticles through ionic bonds [35]. This type of nanocomposite hydrogel can self-heal fractures through chain diffusion and re-binding. We have successfully extended the self-healing model in [37] to explain the healing behavior of nanocomposite hydrogels (Fig. 4A) [36].
Figure 4. (A) Self-healing model of nanoparticle-linked networks[35,36]. (B) Light-activated healing model [38]. (C) Electrically-induced interfacial bonding model[39].
2.3. Effect of external stimuli
The self-healing model can also be used to model stimuli-activated healing. Optically healable polymer is a type of self-healing polymer that harnesses external visible or UV lights to activate the self-healing reaction around the fracture interface [48-50]. We consider that light-triggered free radicals around the healing interface can facilitate the interpenetration of polymer chains, thus establishing two groups of diffusion-reaction systems: light-activated production of free radicals and diffusion-binding of polymer chains. In this way, we can theoretically explain the light-activated healing of polymer networks crosslinked by optically-responsive nanoparticles or organic photophores (Fig. 4B) [38].
2.4. Extend to interfacial bonding of soft materials
Motivated by the pioneering research of Zhao [51,52] and Suo [53-55], the design of tunable/tough bonding between soft materials attracts much attention. We expect that our interfacial self-healing model may be used to provide theoretical explanations for the emerging tough-bonding studies. Taking electrophoresis-induced bonding as an example [39], we consider charged polymers chains are driven by an electric field to move across the interface to interpenetrate into the respective material matrix, and form ionic bonds with chains of opposite charges. A model for the electrically-driven reptation-like motion of polymer chains is formulated. Our theory successfully explains the electrically-induced bonding increase and decrease between charged polymer networks (Fig. 4C) [39].
3. Additive manufacturing of self-healing polymers
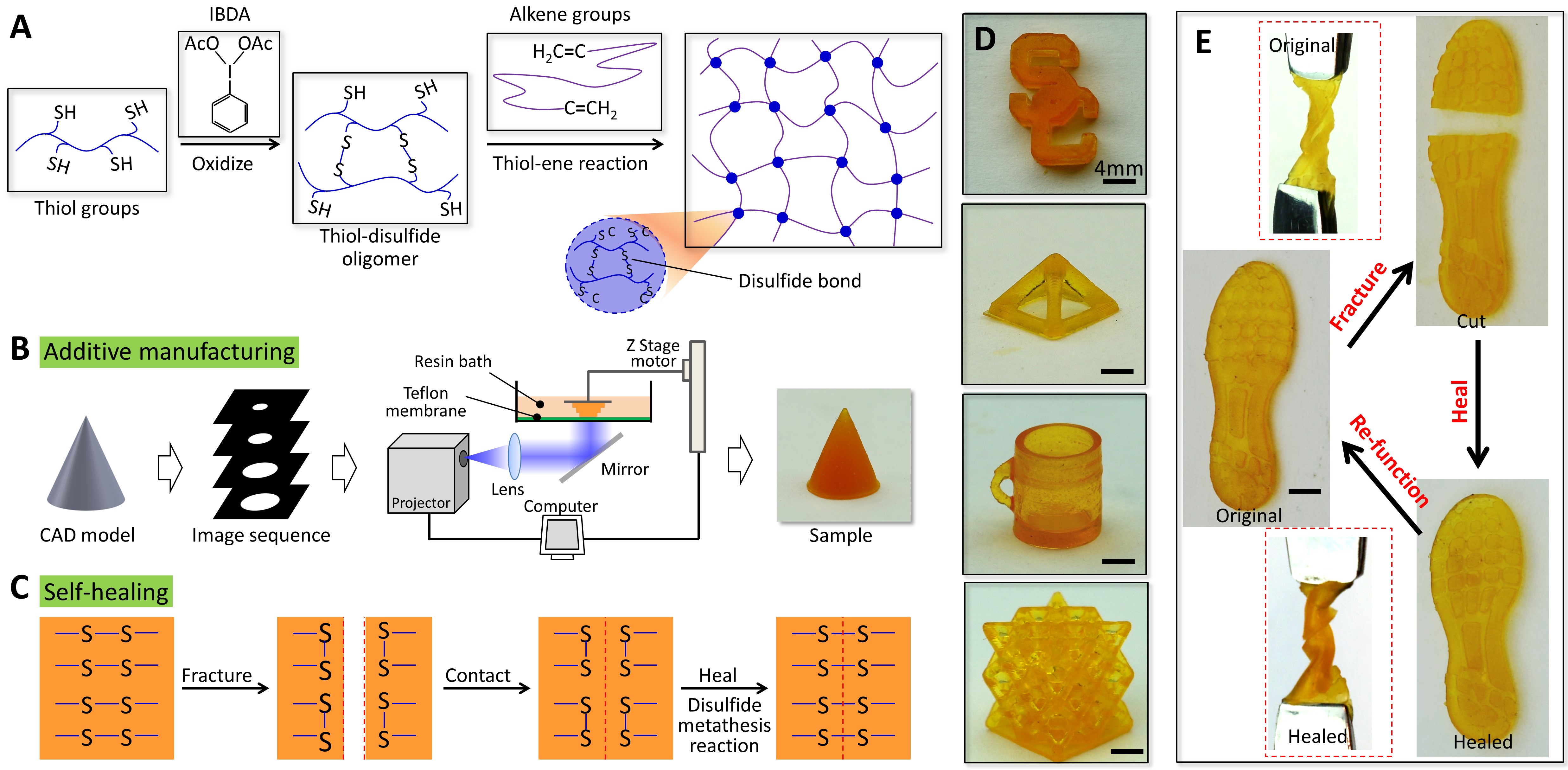
Besides the theoretical effort, we also develop an experimental strategy for photopolymerization-based additive manufacturing (AM) of self-healing elastomers with free-form architectures (Fig. 5) [40]. The strategy relies on a molecularly designed photoelastomer ink with both thiol and disulfide groups, where the former facilitates a thiol-ene photopolymerization during the AM process, and the latter enables a disulfide metathesis reaction during the self-healing process. Using projection microstereolithography systems, we demonstrate rapid AM of single- and multimaterial elastomer structures in various 3D complex geometries within a short time (e.g., 0.6 mm × 15 mm × 15 mm/min=13.5 mm^3/min). The resolution can reach 13.5 µm. These structures can rapidly heal fractures and restore their mechanical strengths to 100%. We also demonstrate additive manufacturing of single- and multimaterial self-healable structures for 3D soft actuators, multiphase composites, and architected electronics [40].
Figure 5. Additive manufacturing of self-healing elastomers. (A) Molecular design of the self-healing elastomer (B) Stereolithography-based additive manufacturing process. (C) Schematics to show the disulfide-bond enabled the self-healing process. (D) The manufactured samples. (E) Self-healing of a shoe pad sample. The healing condition is 2 h at 60°C [40].
4. Challenge and opportunity
The theoretical modeling and additive manufacturing of self-healing polymers highlight a number of challenges and opportunities in the field of solid mechanics, additive manufacturing, and polymer science. We list some of them as follows. Welcome to share your thoughts in the comment box.
4.1. Learn from Fracture Mechanics
We have learned a number of classic terminologies from the Fracture Mechanics class: stress intensity factor, energy release rate, J integral, HRR field, and many more. Questions for our generation: What new concepts and terminologies in the field of Self-Healing Mechanics can we invent for the next generation to follow? How will these new terminologies impact the emerging engineering practice and the solid mechanics field?
Related Journal Club: Aug 2017, Fracture mechanics of soft dissipative materials, Rong Long (UC Boulder)
4.2. Guide design of novel self-healing polymers
Emerging self-healing polymers are becoming tougher, quicker, and smarter [5-9]. The existing field of self-healing polymers primarily relies on chemical innovations. Questions for mechanicians: How to harness emerging self-healing models to guide the design of novel self-healing polymers? How to theoretically understand new polymer network architectures, new dynamic bonds, and new triggering stimuli?
Related Journal Club: Jan 2018, Recent advances in liquid crystal elastomers, Shengqiang Cai (UCSD)
Related Journal Club: Mar 2019, Fatigue of hydrogels, Ruobing Bai (Caltech)
4.3. Guide design of interfacial bonding of soft materials
The tough bonding between similar or dissimilar soft materials has been enabling broad applications [51-55]; however, the theoretical understanding has been left behind. Questions for mechanicians: How to harness the emerging diffusion-reaction models of polymer chains to guide the design of novel tunable/tough bonding of soft materials?
Related Journal Club: Dec 2018, Bonding hydrophilic and hydrophobic soft materials for functional soft devices, Qihan Liu (Harvard)
4.4. Harness architectures of self-healing structures
The emerging additive manufacturing technology brings unprecedented architectures to self-healing polymers. The interaction between self-healing and architecture may enable possibilities for broad applications, such as flexible electronics, robotics, biomedical devices, energy storage devices, and lightweight structures.
Related Journal Club: Feb 2018, HASEL artificial muscles for high-speed, electrically powered, self-healing soft robots, Christoph Keplinger (UC Boulder)
Related Journal Club: Mar 2017, Architected materials, Sung Hoon Kang (JHU)
Reference
[1]B. C. Tee et al., Nat. Nanotechnol. 7, 825 (2012).
[2]C. Wang et al., Nat. Chem. 5, 1042 (2013).
[3]A. B. Brochu, S. L. Craig, and W. M. Reichert, J. Biomed. Mater. Res. A 96, 492 (2011).
[4]S. Terryn et al., Sci. Robot. 2, eaan4268 (2017).
[5]Y. Yang and M. W. Urban, Chem. Soc. Rev. 42, 7446 (2013).
[6]Z. Wei et al., Chem. Soc. Rev. 43, 8114 (2014).
[7]V. K. Thakur and M. R. Kessler, Polymer 69, 369 (2015).
[8]D. Y. Wu, S. Meure, and D. Solomon, Prog. Polym. Sci. 33, 479 (2008).
[9]W. H. Binder, Self-healing polymers: from principles to applications (John Wiley & Sons, 2013).
[10]S. R. White et al., Nature 409, 794 (2001).
[11]K. S. Toohey et al., Nat. Mater. 6, 581 (2007).
[12]X. Chen et al., Science 295, 1698 (2002).
[13]B. Ghosh and M. W. Urban, Science 323, 1458 (2009).
[14]K. Imato et al., Angew. Chem. Int. Ed. 51, 1138 (2012).
[15]Y.-X. Lu and Z. Guan, J. Am. Chem. Soc. 134, 14226 (2012).
[16]P. Cordier et al., Nature 451, 977 (2008).
[17]Y. Chen et al., Nat. Chem. 4, 467 (2012).
[18]Q. Wang et al., Nature 463, 339 (2010).
[19]T. L. Sun et al., Nat. Mater. 12, 932 (2013).
[20]R. Wool and K. O’connor, J. Appl. Phys. 52, 5953 (1981).
[21]R. P. Wool, Soft Matter 4, 400 (2008).
[22]E. B. Stukalin et al., Macromolecules 46, 7525 (2013).
[23]A. C. Balazs, Mater. Today 10, 18 (2007).
[24]R. Long et al., Macromolecules 47, 7243 (2014).
[25]C.-Y. Hui and R. Long, Soft Matter 8, 8209 (2012).
[26]K. Yu et al., J. Mech. Phys. Solids. 94, 1 (2016).
[27]R. A. Bilodeau and R. K. Kramer, Frontiers in Robotics and AI 4, 48 (2017).
[28]A. R. Studart, Chem. Soc. Rev. 45, 359 (2016).
[29]U. G. Wegst et al., Nat. Mater. 14, 23 (2015).
[30]S. J. Benight et al., Prog. Polym. Sci. 38, 1961 (2013).
[31]Z. Zou et al., Sci. Adv. 4, eaaq0508 (2018).
[32]S. Liu and L. Li, ACS Appl. Mater. Interfaces 9, 26429 (2017).
[33]M. A. Darabi et al., Adv. Mater. 29, 1700533 (2017).
[34]X. Kuang et al., ACS Appl. Mater. Interfaces 10, 7381 (2018).
[35]Q. Wang and Z. Gao, J. Mech. Phys. Solids. 94, 127 (2016).
[36]Q. Wang, Z. Gao, and K. Yu, J. Mech. Phys. Solids. 109, 288 (2017).
[37]K. Yu, A. Xin, and Q. Wang, J. Mech. Phys. Solids. 121, 409 (2018).
[38]K. Yu, A. Xin, and Q. Wang, J. Mech. Phys. Solids. 124, 643 (2019).
[39]A. Xin et al., J. Mech. Phys. Solids. 125, 1 (2019).
[40]K. Yu et al., NPG Asia Mater. 11 (2019).
[41]T. L. Anderson, Fracture mechanics: fundamentals and applications (CRC press, 2017).
[42]Q. Wang et al., J. Mech. Phys. Solids. 82, 320 (2015).
[43]G. I. Bell, Science 200, 618 (1978).
[44]P. G. de Gennes, J. Chem. Phys. 55, 572 (1971).
[45]P.-G. De Gennes, Scaling concepts in polymer physics (Cornell university press, 1979).
[46]A. B. Ihsan et al., Macromolecules 49, 4245 (2016).
[47]X. Zhao, Soft Matter 10, 672 (2014).
[48]G. L. Fiore, S. J. Rowan, and C. Weder, Chem. Soc. Rev. 42, 7278 (2013).
[49]Y. Amamoto et al., Adv. Mater. 24, 3975 (2012).
[50]M. Burnworth et al., Nature 472, 334 (2011).
[51]H. Yuk et al., Nat. Commun. 7, 12028 (2016).
[52]H. Yuk et al., Nat. Mater. 15, 190 (2016).
[53]Y. Gao, K. Wu, and Z. Suo, Adv. Mater. 31, 1806948 (2019).
[54]J. Yang, R. Bai, and Z. Suo, Adv. Mater. 30, 1800671 (2018).
[55]Q. Liu et al., Nat. Commun. 9, 846 (2018).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Good review
Dear Qiming,
Thank you for the wonderful review. I studied nanocomposite hydrogel before, so I am very interested in the self-healing hydrogels. I have a few questions and comments and wish to hear your perspective.
(1) Many self-healing hydrogels are very soft, with a Young's modulus 1~dozens of kPa[1-2]. Can high-modulus hydrogels self-heal themselves? When the chains are long in soft hydrogels, it is easy for them to reconnect; while when the chains are short in stiff hydrogels, the network may constrain them to reconnect after fractured. Is this picture right?
(2) As you mentioned, most researchers use tensile strength as a benchmark to evaluate the self-healing capability of one material, is it possible to use the fracture energy as the parameter to evaluate? In this way, people may cut a crack in the specimen and measure the fracture energy before/after the healing.
(3) We just published a paper on the strong adhesion in the 3D printing of heteregeneous soft materials [3]. It may be possible to extend this technology to the self-healing bimaterials with strong adhesion.
[1] J. Insu, J.X.Cui, W.R. Illeperuma,J. Aizenberg, J. Vlassak. Adv.Mater. 28, 2016.
[2] K.Haraguchi et al, Macromol. Rapid Commun. 32, 2011.
[3] H. Yang et al, Adv.Funct. Mater.2019, 1901721.
In reply to Good review by Tang jingda
Re: Good review
Dear Jingda,
Thank you for your interest.
(1) Stiff polymers/gels can also be self-healable. Here are two examples: [1-2]. You are right that the mobility of polymer chain is constrained by the matrix within these materials; therefore, external intervention is required to increase the chain mobility during the healing process, for example, heating, lighting, or applying catalyst.
(2) You have a great point. Most of the existing researchers in the field of self-healing materials use tensile strength as the indicator for the healing performance. Tensile strength can be a good indicator only when the sample number is large enough to eliminate the effect of material defects. Fracture toughness is a better indicator but the measurement is more challenging.
In terms of theoretical understanding, the fracture toughness should include two parts: intrinsic fracture energy due to dissociating or fracturing crossover chains and dissipating fracture energy due to the loading-unloading processing zone. Xuanhe and Rong did fantastic jobs in elucidating the mechanism of these two parts within the framework of fracture of soft materials [3-4]. To the best of my understanding, the current models are still semi-analytical as the processing zone is highly non-linear. I definitely expect the existing theories to be extended to understand the fracture energy of self-healing polymers. We are actually working on a theory to bridge the gap between the tensile strength and the fracture energy.
(3) Congratulations on your great work! According to Xuanhe's work [5], strong bonding/adhesion requires both strong bridges and tough matrix. Involving tough matrices may also be an important factor. Look forward to your future exciting work!
Best regards,
Qiming
Reference
[1] M. Burnworth et al., Nature 472, 334 (2011).
[2] Y. Yanagisawa et al., Science 359, 72 (2018).
[3] T. Zhang et al., Extreme Mechanics Letters 4, 1 (2015).
[4] Y. Qi, J. Caillard, and R. Long, J. Mech. Phys. Solids. 118, 341 (2018).
[5] H. Yuk et al., Nat. Mater. 15, 190 (2016).
Assembling self-healing polymer building blocks
Dear Qiming,
Thanks for your very nice review. The self-healing performances of these polymers are really impressive. You already showed very promising methods of making 3D structures with self-healing polymers based on additive manufacturing. It seems still challenging to make large structures with additive manufacturing (3D printing), in terms of time and cost. By leveraging the self-healing properties, I was wondering whether it is possible to assemble small building blocks of self-healing polymers into a large structure (like building Lego) and then let the assembled structure heal to form a continuous 3D structure?
Best,
Teng
In reply to Assembling self-healing polymer building blocks by Teng zhang
Re: Assembling self-healing polymer building blocks
Dear Teng,
Yes, I guess. The only requirement is that the self-healing material should be stiff enough. Here are two papers doing so, yet not with self-healing materials:
Cheung, Kenneth C., and Neil Gershenfeld. "Reversibly assembled cellular composite materials." science 341.6151 (2013): 1219-1221.
Dong, Liang, Vikram Deshpande, and Haydn Wadley. "Mechanical response of Ti–6Al–4V octet-truss lattice structures." International Journal of Solids and Structures 60 (2015): 107-124.
It is really a tradeoff: slower for bigger structures or faster for smaller structures. It is a long-lasting debate in the field of additive manufacturing. That is also why there is an acute demand for large-scale additive manufacturing systems.
Best regards,
Qiming
Dear Qiming,
Dear Qiming,
Thank you for this very informative and fantastic review on self-healing mechanics. I am particularly interested in the mechanism of self-healing:
(1) First of all, what determines the healing time? As evident in your Fig.3, some materials with dynamic covalent bonds heal themselves in 2-3 hours (Fig. 3B), while it takes 24 hours (Fig. 3A) for some other self-healing materials with the same bond type to fully recover its initial mechanical behavior. Why is that?
(2) Fig. 3C shows materials with hydrogen bonds heal in about 2-5 hours. Intriguingly, recent works found that nano-papers or densified woods which contain a high density of hydrogen bonds did not show obvious self-healing behavior (Processing bulk natural wood into a high-performance structural material, Nature, 554, 224-228 (2018); Anomalous scaling law of strength and toughness of cellulose nanopaper). Could you please shed some insights on this?
(2) Congrats on your great papers examining the self-healing behavior due to dynamic bonds. I wonder if the model can be also applied to decipher the self-healing via chain entanglement?
(3) It is known that the famous double-network hydrogel composed of polyacrylamide and alginate can recover its initial stress-strain response to some extent (not 100%) after a day. Can we also define it as a self-healing hydrogel?
Best regards,
Zheng
In reply to Dear Qiming, by Zheng Jia
Reply to Zheng Jia
Dear Zheng,
Thanks for your interest.
(1) The self-healing time is determined by two factors: chain diffusion and bond kinetics. With a similar bond type, we may assume they have similar bond kinetics; then the difference primarily comes from the chain diffusion. The chain diffusion is governed by two factors: chain length and friction coefficient. The chain diffusivity increases with decreasing chain lengths. The friction is mainly governed by the matrix character; for example, hydrogels may have smaller chain frictions, but stiff polymers may feature much higher chain frictions.
(2) I guess it is because of the low mobility of backbone structures of nanopapers and woods. The healing process requires the hydrogen-bond-anchered chains to flexibly diffuse and penetrate into the other matrix.
(3) I guess you may. Here I consider the healing process as a coupling of chain diffusion and bond reaction. If you consider the chain entanglement, you may not need to consider the bond reaction. Also, I need to point out that the healing physical picture due to chain entanglement in polymer melts has been proposed since 1980s. Here are some references:
[20]R. Wool and K. O’connor, J. Appl. Phys. 52, 5953 (1981).
[21]R. P. Wool, Soft Matter 4, 400 (2008).
(4) The partial healing is due to re-association of ionic bonds, while covalent bonds are permanently broken. There is no standard definition for self-healing polymer networks. Some researchers may still call particle self-healing as "self-healing". Here is an example from Dr. David Weitz group:
Wu, Jinrong, Li‐Heng Cai, and David A. Weitz. "Tough Self‐Healing Elastomers by Molecular Enforced Integration of Covalent and Reversible Networks." Advanced Materials 29.38 (2017): 1702616.
Best regards,
Qiming
Healing time and shape recovery
Dear Qiming,
Thank you for sharing with us many exciting thoughts on the self-healing soft materials. I have two questions on which I hope you could share some thoughts.
1. Following Zheng's question, my impression of most self-healing materials is that many self-healing materials that can heal fast are typically soft and brittle, while self-healing materials that are mechanically robust often heal more slowly. Is this true? If so, is there any way to resolve this conflict?
2. I think another important feature required for many self-healing materials is the capability of shape recovery after healing. For example, a silly putty may heal immediately after fractured into two pieces, but it cannot recover its original shape. To enable this shape recovery, there must be some "memory" in the material of the original configuration, such as an elastic network that provides elasticity and motivates shape recovery. However, an elastic network made of C-C bonds is not healable anymore. A polyampholyte hydrogel by Prof. Jian Ping Gong's group [1] is both self-healing and capable of shape recovery due to the nonuniform distribution of bond strength in the network. How to theoretically or experimentally quantify this feature, and furthermore optimize the overall performance in a general self-healing material?
Thank you,
Ruobing
[1] Sun, T. L., Kurokawa, T., Kuroda, S., Ihsan, A. B., Akasaki, T., Sato, K., ... & Gong, J. P. (2013). Physical hydrogels composed of polyampholytes demonstrate high toughness and viscoelasticity. Nature materials, 12(10), 932.
In reply to Healing time and shape recovery by Ruobing Bai
Re: Healing time and shape recovery
Dear Ruobing,
Thank you for your interest.
1. The material robustness may be roughly indicated by the stretchability of the material. Higher stretchability typically corresponds to longer polymer chains, which in turn leads to smaller chain diffusivity during the healing process. Under this logic, yes, we may roughly conclude that soft materials with better material robustness may heal more slowly. This point can also roughly verified by the data shown in Fig. 3. I guess one way to resolve this is to integrate multiple networks with different chain lengths. That is to say, using the short chains to dissipate energy and restore most of the strength, and using long chains to back the stretchability. The requirement is to involve non-covalent bonds in different networks.
2. Let me first try to understand what you mean here. Correct me if I understand incorrectly. I guess the shape-recovery capability is usually described as "resilience", meaning the capability of returning to its original shape after deformation. Quantitatively, it is described using the damping ratio, that is the ratio between the loss modulus and storage modulus. This is also a term to characterize the viscoelastic property. If the damping ratio is small, the material is very resilience, meaning that the shape immediately returns back to the original state once the applied load is relaxed. Self-healing polymers with non-covalent dynamic bonds usually feature large damping ratios because the dynamic bonds would easily break under deformation; thus, these polymers typically feature poorlow shape resilience.
Let us go back to Gong's paper: their gels are linked by two types of ionic bonds: strong ones and weak ones. From my understanding, the good shape resilience comes from the networks with strong ionic bonds which hold the shape and the weak ionic bonds break to dissipate the energy.
To answer your question regarding "How to theoretically or experimentally quantify this feature", the feature can be quantified using the term "damping ratio" as described above. In terms of theoretical modeling of the damping ratio, we may need to involve a formulation of viscoelastic theory within the time domain and integrate the bond-kinetics of the dynamic bonds in a certain loading rate. I believe this theoretical modeling can be done.
BTW, what you are talking here should not be related to the shape-memory effect of shape-memory polymers.
Hope these can answer your questions.
Best regards,
Qiming